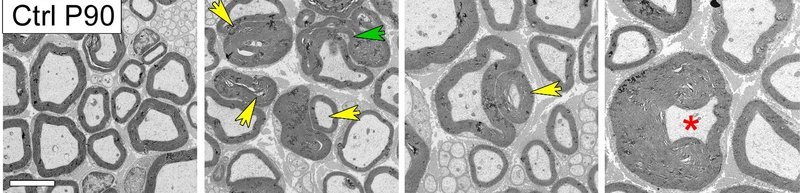
Bild: Alessandra Bolino, IRCCS Ospedale San Raffaele
News • Molekularer Mechanismus entschlüsselt
Charcot-Marie-Tooth: Forscher knacken weiteres Rätsel
Morbus Charcot-Marie-Tooth (CMT) ist die häufigste Form von erblichen Neuropathien. Durch eine genetische Mutation wird die isolierende Myelinschicht der peripheren Nerven nach und nach geschädigt, was beispielsweise beim CMT-Typ 4B zu schwersten Behinderungen führt.
Da die molekularen Grundlagen weitgehend unbekannt sind, ist dieser CMT-Typ bis heute weder behandelbar noch heilbar. Doch jetzt konnten Wissenschaftler vom Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP) in Berlin gemeinsam mit Kollegen aus Mailand, Paris und Mexiko einen neuen molekularen Mechanismus aufzeigen: Demnach kommt dem Protein Rab35 und dem von diesem regulierten mTOR-Signalweg eine zentrale Rolle bei der Myelinscheidenformation im peripheren Nervensystem zu. Erste in-Vivo-Versuche zeigen, dass sich aus den Erkenntnissen neue Therapien ableiten lassen.
Die Arbeit ist jetzt im Fachjournal „Nature Communications“ erschienen.
Dieser Artikel könnte Sie auch interessieren

News • Morbus Charcot-Marie-Tooth
CMT: Unerwarteter Helfer gegen tödliche Nervenkrankheit
Mit rund zwei Millionen Betroffenen auf der ganzen Welt ist Morbus Charcot-Marie-Tooth (CMT) die häufigste erbliche Erkrankung des peripheren Nervensystems, allein in Deutschland sind etwa 30.000 Fälle bekannt. Bislang galt die Krankheit als unheilbar, doch nun haben Forscher in Göttingen eine mögliche Therapie für CMT entdeckt: Lecithin könnte neue Behandlungsoptionen eröffnen.
Die Myelinschicht, die unsere Nervenbahnen umhüllt, sorgt dafür, dass Signale vom Gehirn blitzschnell an Muskeln und Organe weitergeleitet werden. CMT ruft jedoch Defekte in der Myelinisierung vor und stört so die Signalweiterleitung. Einen besonders frühen Krankheitsbeginn zeigt der CMT-Typ 4B; Betroffene sind oft schon im Teenageralter auf den Rollstuhl angewiesen. Im schlimmsten Fall dehnt sich die Nervendegeneration auf die Atemwege aus und es kann zum Tod durch Atemstillstand kommen. Momentan gibt es keinerlei Aussicht auf Heilung.
Wissenschaftler des Leibniz-Forschungsinstituts für Molekulare Pharmakologie (FMP) in Berlin haben zusammen mit den Forscherteams um Prof. Alessandra Bolino (IRCCS Ospedale San Rafaelle Universität, Mailand), Prof. Arnaud Echard (Sorbonne Universität/Institut Pasteur, Paris und Prof. Genaro Patiño-López (Hospital Infantil, Mexiko) die noch weitgehend unbekannten molekularen Mechanismen der Erkrankung näher erforscht. Bei Forschungen zum Protein Rab35 entdeckte das Berliner Team um Linda Sawade und Prof. Volker Haucke mehr oder weniger zufällig, dass diese kleine GTPase, die in der Regulation des intrazellulären Membrantransports involviert ist, mit drei CMT-4B-assoziierten Proteinen interagiert: MTMR2, MTMR5 und MTMR13 funktionieren bei CMT-4B-Patienten aufgrund einer Genmutation nicht richtig oder fehlen ganz. Diese drei kritischen Proteine gehören zur Gruppe der Myotubularin Related (MTMR)-Phosphatidylinositol (PI) Phosphatasen, die spezifisch die endosomalen Signallipide PI(3)P und PI(3,5)P2 an der 3‘-Phosphatgruppe hydrolysieren, also Phosphate von Lipiden abspalten.
Wir vermuten, dass dieser pathologische Vorgang aus einer beeinträchtigten Rekrutierung von MTMR-Komplexen resultiert
Linda Sawade
„Wir konnten in unserer Arbeit zeigen, dass das Protein Rab35 das Längen-Wachstum der Myelinscheide reguliert, indem es die beiden Pseudophosphatasen MTMR13 und MTMR5 bindet und rekrutiert, und dadurch auch die mit ihnen im Komplex gebundene aktive Phosphatase MTMR2“, berichtet Linda Sawade, Erstautorin der Studie. Dass Rab35 diesen gesamten Proteinkomplex bindet und somit eine wichtige Rolle bei der Regulierung der Myelinscheidenformation spielt, war neu. Bestätigt wurde der Fund an Knock-Out Mäusen, denen das Protein spezifisch in Schwann-Zellen fehlte, jene Zellen im peripheren Nervensystem, welche die Myelinscheiden bilden. Das fehlende Rab35-Protein führte zu einer schweren degenerativen Zerstörung der Myelinscheiden des Ischiasnerven.
Parallel dazu beobachteten die Forscher eine abnormal erhöhte Aktivität des mTORC1-Signalweges – einem der zentralen Signalverschaltungskomplexe für die Regulation der Myelinscheidenformation im Nervengewebe. Durch die medikamentöse Hemmung des hyperaktiven Signalkomplexes mit dem Wirkstoff Rapamycin konnten die Nervenschäden in den Knock-out-Mäusen jedoch teilweise wieder rückgängig gemacht werden. Weitere Experimente an kultivierten Zellen, in denen Rab35 unterdrückt wurde, untermauerten die positiven Effekte einer mTORC1-Inhibition auf die geschädigten Myelinsegmente. Zudem konnten die Forscher aus der Abwesenheit des Rab35 Proteins einen wichtigen Rückschluss ziehen: mTORC1 ist hyperaktiv, weil PI(3)-Phosphate nicht mehr reguliert werden, es kommt zu einer Akkumulation von PI(3)P und PI(3,5)P2 Lipiden. „Wir vermuten, dass dieser pathologische Vorgang aus einer beeinträchtigten Rekrutierung von MTMR-Komplexen resultiert“, erläutert die Biochemikerin und Zellbiologin Linda Sawade. „Das würde im Umkehrschluss bedeuten, dass Rab35 die Aktivität von mTORC1 normalerweise unterdrückt, in dem es MTMR-Phosphatasen zu Lysosomen rekrutiert.“
Kurz zusammengefasst bedeuten die Ergebnisse für die Grundlagenforschung: Rab35 ist ein bisher unentdeckter Regulator der Myelinscheidenformation im peripheren Nervensystem und ein Repressor von mTORC1. Für CMT4B-Patienten haben die Ergebnisse eine hoffnungsvolle Bedeutung: Die therapeutische Behandlung mit mTORC1-inhibierenden Medikamenten wie Rapamycin könnten zu einer Verbesserung ihres Krankheitsverlaufs führen. Es wäre die erste Behandlungsoption für diese schwere Erkrankung. Arbeitsgruppenleiter Prof. Dr. Volker Haucke: „Mit unserer Arbeit haben wir einen neuen molekularen Mechanismus von einer besonders schweren Form der erblichen Neuropathie aufgedeckt, der klinisch hoch relevant ist und den wir mit dem Mailänder Team um Alessandra Bolino nun noch tiefergehender erforschen wollen.“
Quelle: Leibniz-Forschungsinstitut für Molekulare Pharmakologie (FMP)
05.06.2020