News • Entstehungsmechanismus identifiziert
Lungenfibrose: Neue Möglichkeiten für zielgerichtete Therapien
Forschungsgruppen an der Charité – Universitätsmedizin Berlin und in Heidelberg ist es gelungen, die Entstehung von Lungenfibrose im Detail nachzuverfolgen.
Sie konnten zeigen, dass dem Protein NEDD4-2 eine Schlüsselfunktion für die gesunde Lunge zukommt, und ein Fehlen dieses zentralen Regulators für verschiedene Prozesse bei der Krankheitsentstehung von Bedeutung ist. Wie genau sich die Lungenfibrose entwickelt und wie sie verläuft, lässt sich nun noch besser untersuchen. Auf Grundlage dieser Erkenntnisse können neue Therapieansätze entwickelt werden, wie jetzt im Fachmagazin Nature Communications beschrieben ist.
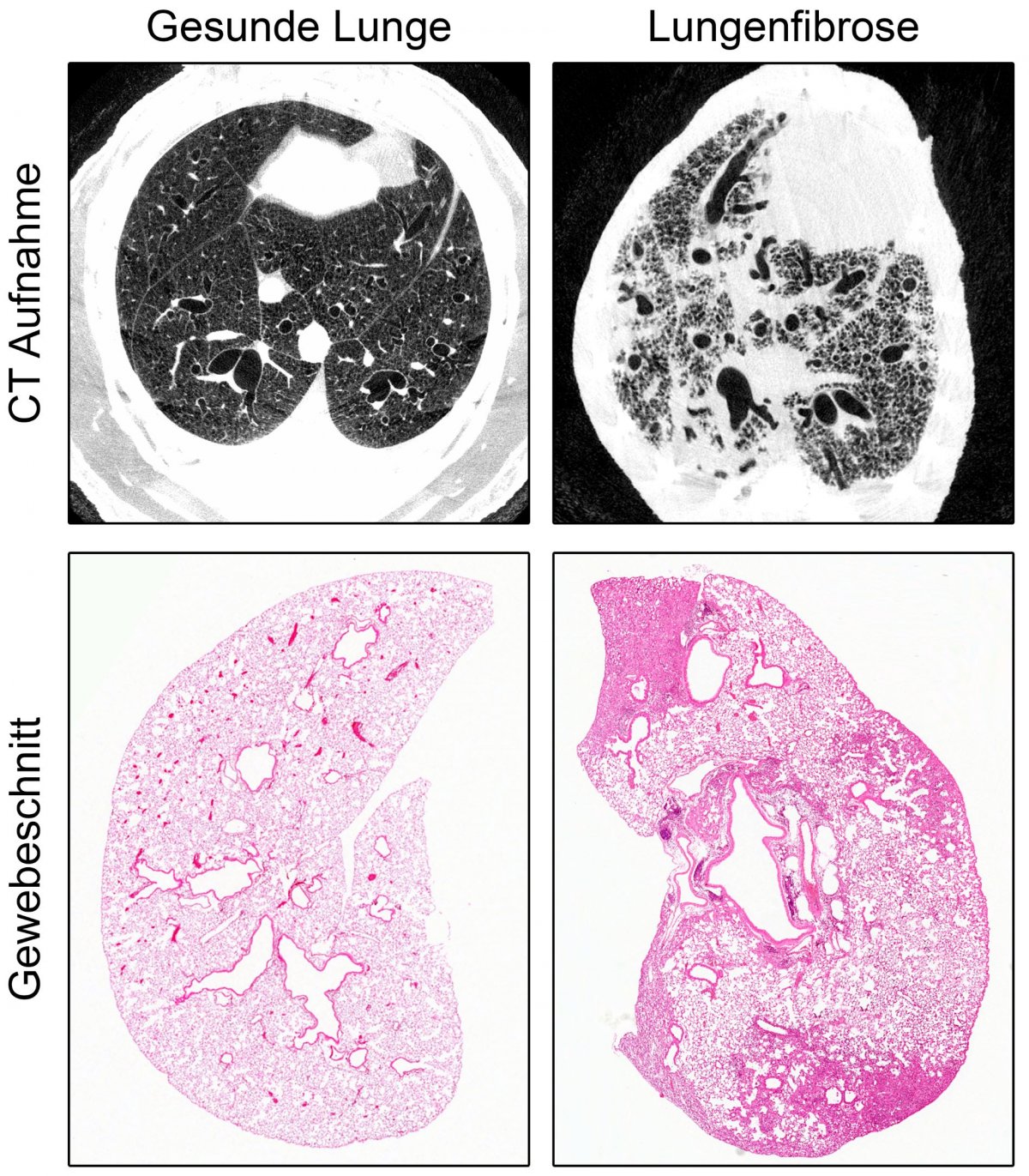
Foto: Leitz/Charité
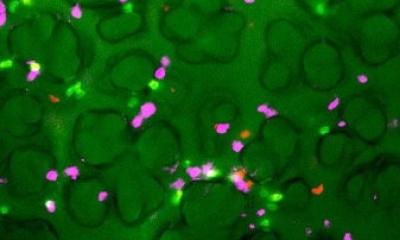
Eine Fibrose der Lunge ist eine schwerwiegende Erkrankung, die vor allem ältere Menschen betrifft und für die es kaum Behandlungsmöglichkeiten gibt. Das Lungengewebe verändert sich zunehmend und vernarbt. Die Ursachen einer Lungenfibrose sind jedoch weitgehend unbekannt und der Mechanismus auf zellulärer Ebene kaum verstanden. Mit einem als „mukoziliäre Clearance“ bezeichneten Selbstreinigungsmechanismus transportieren die auskleidenden Epithelzellen der Lungenschleimhaut mit ihren Flimmerhärchen Bronchialschleim zusammen mit eingeatmeten Erregern und Schadstoffen aus den Atemwegen heraus. Es ist bekannt, dass eine übermäßige Produktion oder ein gestörter Abtransport von Schleim und dessen Bestandteilen, so genannten Mucinen, mit einer Veränderung dieser Epithelzellen einhergeht. Das Protein NEDD4-2 ist am Abbau verschiedener anderer Proteine beteiligt, die durch solche Prozesse die Funktion von Epithelzellen der Lunge regulieren. Das macht NEDD4-2 zu einem zentralen Schlüsselprotein beim Entstehen der Lungenfibrose.
Dem Team um Prof. Dr. Marcus Mall, Direktor der Klinik für Pädiatrie mit Schwerpunkt Pneumologie, Immunologie und Intensivmedizin der Charité und Professor des Berlin Institute of Health (BIH), ist es zusammen mit Forschenden des Deutschen Zentrums für Lungenforschung (DZL), des Universitätsklinikums Heidelberg und des Deutschen Krebsforschungszentrums nun erstmals gelungen, ein Tiermodell zu entwickeln, das die sogenannte idiopathische pulmonale Fibrose (IPF) detailliert widerspiegelt. Da NEDD4-2 für die frühe Entwicklung unverzichtbar ist, wurde das kodierende Gen erst bei erwachsenen Tieren gezielt in der Lunge entfernt. Die Wissenschaftler und Wissenschaftlerinnen untersuchten diese dann zu einem späten Zeitpunkt, der etwa der Diagnosestellung beim Patienten entspricht. Dabei zeigten Messungen der Sauerstoffsättigung eine für die Erkrankung typische Verschlechterung der Lungenfunktion. Durch Gewebeschnitte und Bildgebung der Lunge mittels Computertomographie ließen sich außerdem die strukturellen Kennzeichen einer Fibrose wie eine fleckige Vernarbung nachweisen. Die Bedeutung von NEDD4-2 bei der Krankheitsentstehung von IPF zeigt sich auch daran, dass sowohl die Transkript- als auch die Proteinmenge in Lungenbiopsien von Patienten stark reduziert ist. Außerdem ergab eine Untersuchung des Proteomprofils, also der Gesamtheit aller Proteine, mittels Massenspektrometrie eine hohe Übereinstimmung an Proteinen, die sowohl bei Patienten mit IPF als auch im Tiermodell eine veränderte Expression aufweisen. „Unsere Erkenntnisse können dazu beitragen, die Entstehung und den Verlauf der Lungenerkrankung weiter zu untersuchen und neue Therapien zu entwickeln, beispielsweise können Substanzen, die für eine Therapie infrage kommen, in einem präklinischen Stadium erprobt oder eine Früherkennung der Erkrankung ermöglicht werden“, sagt Prof. Mall.
Dieser Artikel könnte Sie auch interessieren

Artikel • Präzisionsdiagnostik
Künstliche Intelligenz zur Diagnose von Lungenerkrankungen
Unter dem Begriff diffuse parenchymatöse Lungenerkrankungen (DPLD) wird eine Gruppe von mehr als 200 Erkrankungen zusammengefasst, die vom Alveolarepithel, dem Endothel der Lungenkapillaren oder dem pulmonalen Interstitium der Lunge ausgehen. Die große Zahl an Erkrankungen, verbunden mit der Seltenheit der einzelnen Entitäten und deren Vielzahl an Manifestationsformen, stellt Pneumologen,…
Die Forschenden konnten bei der Untersuchung der zugrundeliegenden Krankheitsmechanismen feststellen, dass durch das Fehlen des Proteins NEDD4-2 Epithelzellen in den Atemwegen umgestaltet werden: Der Anteil der verschiedenen Zelltypen ist verändert und die Zellen produzieren zudem größere Mengen bestimmter Mucine. Zusammen mit veränderten Natriumströmen in Epithelzellen und einem dadurch verringerten Volumen des Flüssigkeitsfilms führt dies zu einem gestörten Abtransport – der Selbstreinigungsprozess der Atemwege ist eingeschränkt. Darüber hinaus verursacht das Fehlen von NEDD4-2 eine verstärkte Aktivität des pro-fibrotischen TGFβ-Signalwegs. „Wir konnten somit einen direkten Zusammenhang zwischen dem Fehlen von NEDD4-2 und einer gestörten mukoziliären Clearance sowie der Fehlregulierung des TGFβ-Signalwegs herstellen, zwei Störungen, die nach aktuellen Erkenntnissen in der Pathogenese von IPF eine Rolle spielen“, resümiert Dr. Julia Dürr, Erstautorin der Studie.
In der Behandlung der Lungenfibrose werden bereits seit einigen Jahren anti-fibrotische Therapien eingesetzt. Diese führen zwar meist zu einer Verlangsamung des Vernarbungsprozesses, können eine Lungentransplantation als letzte Behandlungsoption jedoch nicht gänzlich ersetzen. „Eine solche Verlangsamung des Krankheitsverlaufs, aber keine vollständige Heilung konnten wir in der Anwendung eines bereits zugelassenen anti-fibrotischen Medikament in unserem Modell bestätigen." Prof. Mall fügt hinzu: „Wir hoffen nun durch eine verbesserte präklinische Testung dazu beizutragen, dass Therapieansätze schneller entwickelt werden können.“ In weiteren Schritten sollen Biomarker zur Früherkennung getestet sowie potenzielle neue Substanzen zur Behandlung der Lungenfibrose auf ihre Wirksamkeit hin untersucht werden.
Quelle: Charité – Universitätsmedizin Berlin
27.04.2020