


Quelle: shutterstock/963 Creation
Artikel • Medizinprodukte-Verordnung
Tsunami in Sicht? Die MDR und ihre Folgen
Nach sieben Jahren „Bauzeit“ ist die EU-Medizinprodukteverordnung 2017/745 – Medical Device Regulation, kurz MDR am 26.05.2021 verbindlich in Kraft getreten. Pandemiebedingt, aber auch weil diverse Rechtsvorschriften noch nicht finalisiert waren, wurde der eigentlich geplante Geltungsbeginn 2020 um ein Jahr verschoben. Die 12 Monate Aufschub haben aber offensichtlich nicht ausgereicht, das System lauffähig zu machen.
Report: Julia Geulen
Die Komplexität des neuen Systems mit seinen zahlreichen Regularien und Dokumentationspflichten wurde offenbar massiv unterschätzt. So das Fazit des Bundesverbandes Medizintechnologie, BVMed. Denn die neue Verordnung ist nicht praxistauglich: Zu wenige Benannte Stellen, zu viele bürokratische Vorschriften, die vor allem die klein- und mittelständischen Unternehmen stark belasten. Pragmatische Lösungen und eine Verlängerung der Fristen – so die Forderungen der BVMed.
Vorrangiges Ziel der MDR – und da sind sich Industrie, Anwender und Gesetzgeber einig – ist die Gewährleistung der Patientenversorgung mit sicheren Medizinprodukten. Der Weg dorthin ist jedoch umstritten. Im Mittelpunkt steht dabei die Frage, ob Aufwand und Nutzen im richtigen Verhältnis stehen. Denn Files, die im Rahmen der neuen MDR angelegt werden, haben einen gut zehnfachen Umfang, das betrifft nicht nur die Dokumentation, sondern auch die Fülle an zusätzlich geforderten Tests, Prüfverfahren Gutachten und klinischen Untersuchungen.
Dr. Meinard Lugan, Vorstandsvorsitzender der BVMed und Vorstandsmitglied B. Braun Melsungen AG und Marc D. Michel, Vizevorsitzender der BVMed und Geschäftsführer der Peter Brehm GmbH, kritisieren vor diesem Hintergrund eine fehlende Differenzierung. Denn die hohen Zulassungsanforderungen gelten gleichermaßen für neue Produkte, wie auch für Produkte, die schon lange und erfolgreich in der Anwendung sind. Es handelt sich keineswegs um ein speziell deutsches Problem. Ebenso betroffen sind zum Beispiel Italien, Frankreich und Irland, also Länder, in denen die Medizintechnik großgeschrieben wird. Der Druck ist allgemein groß.
Black Box „Benannte Stellen“
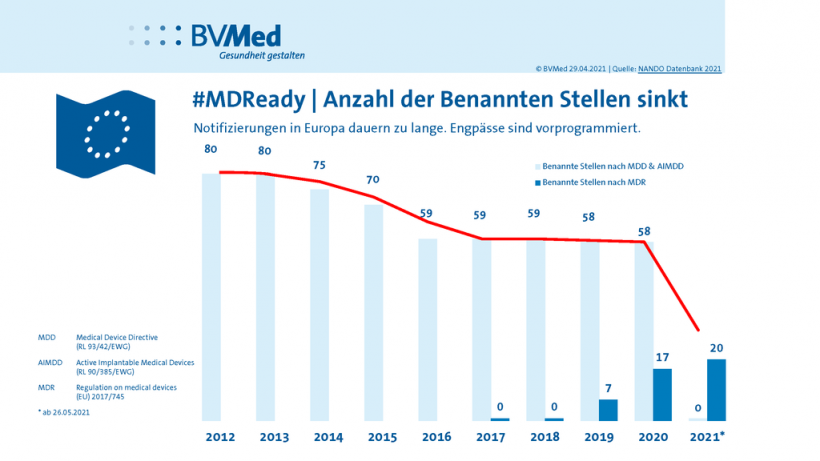
Die Benannten Stellen (Notified Bodies) sind ein Nadelöhr, durch das ein gewaltiger Bearbeitungsstau verursacht wird. Was sind die Gründe? Punkt eins: Der Prozess der Qualifizierung von dringend benötigten zusätzlichen Benannten Stellen vollzieht sich imSchneckentempo. Punkt zwei: Für die hohen Bearbeitungsanforderungen der MDR benötigen die Benannten Stellen sehr viel mehr Prüfzeit pro Antrag. Darauf sind sie nicht ausreichend vorbereitet und auch nicht in der Lage, den bevorstehenden Aufwand realistisch abzuschätzen. Punkt drei: Um das immense Arbeitsvolumen bewältigen zu können, müssen die Benannten Stellen ihr qualifiziertes Personal massiv aufstocken. Ingenieure, Prüfer, Qualitätsmanager – diese Berufsgruppen sind auf dem Stellenmarkt sehr gefragt und daher rar.
Das Resultat: Die einreichenden Hersteller haben keine Planungssicherheit: Wo stehen wir in dem Prozess? Was muss eventuell noch nachgereicht werden, an Daten oder Dokumenten? Wann kann mit einer Zulassung gerechnet werden? Darauf gibt es zurzeit keine verlässlichen Antworten. Lugan: „Die Situation ist beängstigend und sieht bei diagnostischen Produkten, Stichwort IVDR ab Juni 2022, noch trüber aus. Dort gibt es gerade mal eine Handvoll zuständiger Stellen und das bei Produkten, denen gerade jetzt eine hohe Relevanz zukommt, die also schnell auf den Markt kommen sollten.“
Quelle: BVMed e.V.
Bestandsgeschäft in Gefahr
Bereits jetzt ist absehbar, dass bis zum Ende der Übergangsfrist für alte Zertifikate 2024, nur sehr wenige Anträge finalisiert werden können. Alles was liegen geblieben ist, verursacht ein weiteres Ansteigen der Bugwelle. Für 81 Zertifikate werden aktuell 12 Monate benötigt. Selbst wenn Routine und wachsende Erfahrung Wirkung zeigen sollten, der Prozess müsste zehnmal schneller werden, um diese Aufgabe zu bewältigen. Ein weiteres Problem: Innovative Produkte hängen bis auf Weiteres in der Warteschleife. Denn zunächst gilt es die Re-Zertifizierung der alten Produkte voranzutreiben, um das Bestandsgeschäft nicht zu gefährden. Erst wenn das gesichert ist, können sich die Unternehmen der Zertifizierung der neuen Produkte widmen.
Besonders betroffen: kleine und mittelständische Unternehmen (KMUs)
93 Prozent aller Unternehmen der Branche gehören dem Klein- und Mittelstand an. Über die Hälfte der Wertschöpfung von MedTech-Produkten, meist Klasse III Produkte, generieren die KMUs. Zwei Drittel aller Erwerbstätigen der Branche arbeiten in diesen Unternehmen. Und doch sind es gerade die KMUs, die von den Regelungen der MDR besonders hart getroffen werden. Michel erläutert, warum: Mittelständische Unternehmen haben klassischerweise Nischenprodukte, also Produkte, die zwar nicht häufig zum Einsatz kommen, die aber bei entsprechender Indikation für die Patienten von sehr großem Nutzen sind.
Wenn Aufwand und Kosten für eine Rezertifizierung eines solchen, längst etablierten Produktes genauso hoch sind, wie für ein Massenprodukt, können sich die Hersteller diesen Zulassungsaufwand bei gleichzeitig geringer Stückzahl und Umsatz langfristig nicht leisten. Die Folge, die Produkte, müssen aus dem Verkehr gezogen werden. „Es ist mir ein Rätsel, wieso Bestandsprodukte, die seit vielen Jahrzehnten beispielsweise in der Endoprothetik erfolgreich am Markt sind, neu zugelassen werden müssen“, wundert sich Michel.
Dieser Artikel könnte Sie auch interessieren
News • EU-Medizinprodukte-Verordnung
Expertengruppe warnt vor Engpässen in Medizinversorgung
Eine Arbeitsgruppe warnt vor Engpässen in der medizinischen Versorgung: Bewährte Medizinprodukte wie EKG-Geräte müssen aufwendige Zulassungsverfahren durchlaufen.
Problem Klinische Daten
Voraussetzung für die Zertifizierung sind klinische Daten. Aber woher nehmen? Die Kliniken spielen hierbei nur ungern mit. Denn zum einen sind sie nicht darauf ausgelegt, dieser Anfragenflut nach klinischen Daten nachzukommen, zum anderen ist das für sie wenig attraktiv. Bestandsprodukte sind für die wissenschaftliche Arbeit meistens uninteressant. Hier müsste massiv investiert werden, um Strukturen zu schaffen und die Studien zur Generierung dieser Daten attraktiver zu machen.
Eine andere Lösung wäre es – zumindest im Bereich Implantate – auf Registerdaten zurückzugreifen. „Aber ohne Zeitverzug wird auch das nicht gehen“, ist Michel überzeugt. Derweil tickt die Uhr und das Fristende 2024 wird schnell näher rücken. Ein zusätzliches Arbeitspaket stellen die sprachlichen Anforderungen der MDR dar. Selbst wenn die Zulassung für den europäischen Raum vorliegt, sind die Hersteller ab sofort verpflichtet, Dokumente, wie Instrumentationsanleitungen, Aufbereitungsanleitungen, Implantate-Pass, Sicherheitsberichte in der jeweiligen Landessprache vorzuhalten.
Auch hier ist abzuwägen, in welchen Ländern sich der zusätzliche Aufwand für die sprachliche Anpassung noch lohnt. Apropos Ausland: In Folge des Brexit haben für UK vorgesehene Produkte einen eigenen Warenkreislauf. Das lohnt sich nur bei hochvoluminösen Produkten. Den multinationalen Konzernen, die größere Ressourcen vorhalten können, spielt das in die Hände, für den Klein- und Mittelstand mit seinen Nischenprodukten stellt dies ein riesiges Problem dar. Eine Verschiebung von heterogenen Strukturen hin zu multinationalen Großkonzernen ist die Folge.
Dieser Artikel könnte Sie auch interessieren

Artikel • HüftImplantatPfannenfräsSimulator
HIPS: Mit VR und Leichtbauroboter Hüft-OPs simulieren
Mehr als 200.000 Menschen erhalten in Deutschland pro Jahr eine Hüftprothese. Um Komplikationen zu vermeiden und die Lebensdauer der Prothese zu verlängern, müssen die Implantate möglichst genau in die Hüftpfanne eingepasst werden. Doch der Eingriff ist schwierig, besonders das sogenannte Ausfräsen des Acetabulums gehört zu den heikelsten Schritten, die sich zudem schwer üben lassen.
Beispiele aus der Endoprothetik
Jedes Jahr werden in Deutschland ca. 180.000 künstliche Hüftgelenke implantiert. Nach einer gewissen Zeit kommt es häufig zu sogenannten Revisions-OPs. Dazu werden hoch spezifische Produkte benötigt, auch in Abhängigkeit von der medizinischen Problematik des Patienten. So ein besonderes Hüftgelenksystem wird bundesweit ungefähr 500 Mal benötigt. Über diese Gruppe von 500 Patienten klinische Daten zu erfassen, ist absurd. Die Statistik ist bei so geringen Zahlen nicht aussagekräftig. Was also tun? Ein echtes Dilemma, denn das Produkt wird von bestimmten Patienten unbedingt benötigt. Andere Verkaufszahlen liegen in Deutschland bei unter 100 Stück, so zum Beispiel ein Produkt, das bei defektem Bandapparat im Knie zum Einsatz kommt. „Ein Produkt, das sehr selten zum Einsatz kommt, für den Patienten aber ein echter Segen ist, weil es ihm schlichtweg die Amputation erspart“, führt Michel aus. „Wir müssen deshalb alles daransetzen, um diese Nischenprodukte im Verkehr zu halten“, so Michel.
Rasante Kostensteigerungen
Der Stundensatz seitens der Benannten Stelle erhöht sich voraussichtlich um 38 Prozent. Die Anforderungen an die Ausbildung des Personals steigen ebenfalls, und mit ihnen die Personalkosten. Die Begründung ist nachvollziehbar, trifft die Hersteller aber dennoch hart. Auch die Kosten für technische Dokumentationen erhöhen sich um 53 Prozent, Tendenz steigend. Für die mittelständischen Betriebe mit kleinen Stückzahlen heißt das: Preisverfall bei gleichzeitig steigenden Kosten für die Rezertifizierung. Gerade Unternehmen und Start Ups, die mit Klasse IIb- und III-Produkten am Markt sind, sind damit über kurz oder lang am Limit. Ein internationaler Vergleich: In den USA kostet die Zulassung eines neuen Produktes durch die FDA, die gleichermaßen über strenge Regularien verfügt, etwa 27.000 EUR. Für ein Klasse III-File in der EU muss mit dem Faktor 2 multipliziert werden, zuzüglich der Kosten, die durch Verzögerungen entstehen. Deutschland und Europa könnten sehr schnell ins Hintertreffen geraten, wenn es um Wettbewerbsfähigkeit und den Zugang für Patienten zu modernen Medizinprodukten geht.
Dieser Artikel könnte Sie auch interessieren

News • EU-Marktzugang
MDR: Schweizer Medtech zum "Drittstaat" degradiert
Der erste Dominostein ist gefallen: Wegen des fehlenden Institutionellen Abkommens (InstA) hat die Europäische Union (EU) das Abkommen über die gegenseitige Anerkennung von Konformitätsbewertungen (Mutual Recognition Agreement, MRA) nicht aktualisiert. Mit dem Geltungsbeginn der neuen EU-Medizinprodukteverordnung (Medical Device Regulation, MDR) verliert die Schweizer Medizintechnikindustrie…
Besorgniserregend
Dies alles wird von den Unternehmen mit großer Sorge betrachtet. “In der Konsequenz könnte der Erstmarkt für Medizinprodukte in der EU wegbrechen und sich in Richtung USA verlagern.“, so Michel. Zuliefererunternehmen, so die Schätzung, werden voraussichtlich 30 Prozent ihres Portfolios abstoßen, 10 Prozent möglicherweise in der nächsten Zeit komplett aufgeben. Medizintechnik ist dann so teuer, dass der Fokus auf andere Felder wie Kunststoff, Autos oder Luftfahrt gelegt wird.
„Es steht eine Bereinigung einer mittelständischen Industriestruktur auf dem Spiel“, befürchtet Michel. „Wir benötigen dringend Unterstützung – vor allem bei der Erhebung von klinischen Daten. Wenn wir das nicht organisiert bekommen, erhalten wir weder die Rezertifizierung von Bestandsprodukten, noch können wir neue Produkte auf den Markt bringen“, legt Michel den Finger in die Wunde.
Was ist zu tun? Die Branche postuliert verschiedene Lösungsansätze. Grundsätzlich muss das Regelwerk der MDR weiterentwickelt werden und zwar unter Berücksichtigung der realen Verhältnisse.
An erster Stelle steht die zügige und merkliche Aufstockung der Benannten Stellen. Diese müssen dann auch in der Lage sein, die verschiedenen Medizinprodukte-Klassen gleichermaßen abzudecken. Anders ist die Bugwelle, die das System langfristig vor sich herschiebt, nicht zu brechen. Die Übergangsphase muss über 2024 hinaus verlängert werden, so dass die angestauten Files sukzessive abgearbeitet werden können und Platz für andere geschafft wird. Wünschenswert wäre es, wie in anderen Ländern bereits erfolgreich geschehen, digitale Audits umzusetzen. Dazu fehlt in Deutschland zurzeit noch der Rechtsrahmen. Auch zum Thema klinische Daten liegen Vorschläge auf dem Tisch: So wäre es sehr hilfreich, wenn zum Beispiel aus den Daten, die Krankenhäusern vorliegen, automatisch die Marktüberwachung hergeleitet werden könnte. Lugan: „Das wäre auch ein Schritt in eine digitale Richtung, aber leider stehen dem im Moment alle rechtlichen Systeme entgegen.“
Weniger Chancen werden der KI eingeräumt, auch wenn europaweit umfangreiche Patientendaten zur Verfügung stehen. Die Option eines Poolings wird aber skeptisch gesehen, zu aufwändig und uneinheitlich sind die Vertragsverhandlungen mit dem Datenschutzbeauftragten auf Landesebene. Eher setzt man auf die möglichst schnelle Nutzung von Registerdaten, etwa dem Endoprothesen-Register. Denn die Register haben in den nächsten Jahren einen hohen Stellenwert, weil abgesehen von Ein- und Ausbau eines Implantats viele andere Parameter zu erheben sind, um Aussagen zum Beispiel für den Sicherheitsbericht generieren zu können.
Niederschwellige Stellschrauben
Eigentlich ist vom Gesetzgeber 2027 ein MDR-Check vorgesehen. Dies ist aus Sicht der Hersteller viel zu spät. Bis dahin wird ein Teil der Branche bereits aufgegeben haben, so die Vermutung. Sinnvoller wäre die selbstkritische Prüfung jetzt vorzunehmen, um den konkreten Handlungsbedarf abzuleiten und umzusetzen. „Fristen verlängern etwa und Klasse 1 Produkte herausnehmen aus dem Prozess – das wäre hilfreich – und zwar bitte nicht erst 2027“, konkretisieren Lugan und Michel. Mit einer grundsätzlichen Gesetzesänderung ist nicht zu rechnen, zu viel Zeit und Aufwand wurde bereits investiert. Aber alle Möglichkeiten unterhalb der Gesetzesebene sollten genutzt werden, um das System doch noch lauffähig zu machen. Mit Schrecken blickt die Branche auf die EU-Verordnung für In-Vitro-Diagnostika (IVDR), die im Juni 2022 verpflichtend in Kraft treten soll. MDR und IVDR gemeinsam könnten sich dann zur Monsterwelle entwickeln.
08.06.2021


