Big Data
Proteinstruktur mittels Big Data vorhergesagt
Ohne Proteine geht im Körper nichts – sie sind die molekularen Alleskönner in unseren Zellen. Arbeiten sie nicht richtig, kann das schwere Krankheiten auslösen wie etwa Alzheimer. Um Methoden zu entwickeln, nicht funktionierende Proteine zu reparieren, muss man deren Struktur kennen. Mit einem Big-Data-Ansatz haben Forscher des Karlsruher Instituts für Technologie (KIT) nun eine Methode entwickelt, mit der sie Proteinstrukturen vorhersagen können.
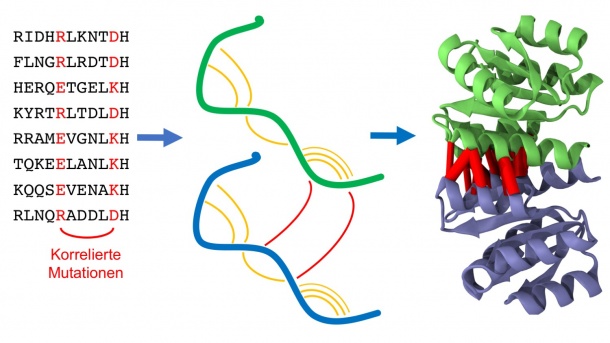
Wie die Forscher in der Zeitschrift Proceedings of the National Academy of Sciences of the United States of America (PNAS) berichten, ist es ihnen gelungen, auf Basis statistischer Analysen auch komplizierteste Proteinstrukturen unabhängig vom Experiment vorherzusagen. Diese experimentell zu bestimmen ist sehr aufwändig und der Erfolg nicht garantiert. Proteine bilden die Grundlage des Lebens. Als Strukturproteine sind sie am Aufbau von Gewebe wie Nägeln oder Haare beteiligt. Andere Proteine arbeiten als Muskeln, steuern den Stoffwechsel und die Immunabwehr oder übernehmen den Sauerstofftransport in den roten Blutkörperchen
Die Grundstruktur von Proteinen mit bestimmten Funktionen ähnelt sich auch bei sehr verschiedenen Organismen „Ob Mensch, Maus, Walfisch oder Bakterium, die Natur erfindet Proteine für verschiedene Lebewesen nicht immer neu, sondern variiert sie lediglich durch evolutionäre Mutation und Selektion“, so Alexander Schug vom Steinbuch Centre for Computing (SCC). Solche Mutationen können beim Auslesen der Erbinformationen, aus denen die Proteine zusammengesetzt sind, leicht identifiziert werden. Treten sie nun paarweise auf, liegen die beteiligten Proteinabschnitte meist räumlich nahe beieinander. Diese einzelnen Informationen vieler räumlich benachbarter Abschnitte können mit einem Computer wie ein großes Puzzle zu einer genauen Vorhersage der dreidimensionalen Struktur zusammengesetzt werden. Denn: „Um die Funktion eines Proteins wirklich im Detail zu verstehen und eventuell auch zu beeinflussen, muss man den Ort jedes einzelnen Atoms kennen“, sagt Schug.
Der gelernte Physiker nutzt in seiner Arbeit einen interdisziplinären Ansatz mit Methoden und Ressourcen der Informatik und Biochemie. Mit Hilfe von Hochleistungsrechnern hat er die frei verfügbaren Erbinformationen tausender verschiedener Organismen von Bakterien bis hin zum Menschen nach in Verbindung stehenden Mutationen durchsucht. „Durch die Kombination von modernster Technik und einem wahren Schatz an Datensätzen konnten wir knapp 2000 verschiedene Proteine untersuchen – das ist eine völlig neue Größenordnung im Vergleich zu bisherigen Studien“, sagt Schug. Dies unterstreiche eindrucksvoll die Leistungsfähigkeit dieser Methodik und verspricht großes Potenzial für breite Anwendungen von der Molekularbiologie bis hin zur Medizin. Auch wenn die gegenwärtige Arbeit Grundlagenforschung sei, wie Schug betont, können die Ergebnisse in Zukunft beispielsweise in neue Behandlungsmethoden von Krankheiten einfließen.
Quelle: KIT
23.03.2017