Bildquelle: Universitätsklinikum Heidelberg
News • Molekulares Profiling in Minuten statt Wochen
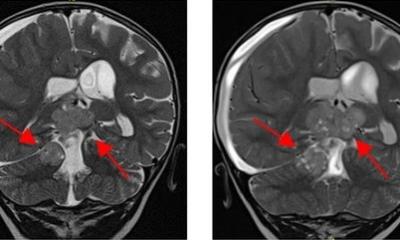
Hirntumor-Analyse im Schnelldurchgang
Wissenschaftler der Medizinischen Fakultät Heidelberg der Universität Heidelberg haben neue Analysemethoden für besondere Herausforderungen der Tumordiagnostik entwickelt:
Eine liefert bereits im begrenzten zeitlichen Rahmen einer Hirn-OP wichtige Informationen für die Einordnung der Tumoren. Die andere könnte mit Hilfe Künstlicher Intelligenz die Genauigkeit der Klassifizierung verbessern, wenn nur wenig Tumormaterial für die Auswertung vorliegt. Die Methodiken beschreiben sie in den Fachjournalen Nature Medicine und Nature Cancer.
Die molekulare Auswertung einer Tumorprobe kann aufgrund der komplexen Analyseverfahren bis zu zwei Wochen dauern. Ein Team der Neuropathologie der Medizinischen Fakultät Heidelberg und des Deutschen Krebsforschungszentrums (DKFZ) haben Analysemethoden entwickelt, die, nachdem das Erbgut aus den Tumorzellen gewonnen wurde, innerhalb von 30 Minuten eine erste Typisierung anhand bestimmter Eigenschaften des Krebsgenoms liefern. Ein umfassendes molekulares Profil der Tumorprobe liegt innerhalb von 24 Stunden vor. Bei Vergleichsanalysen von 78.000 archivierten Tumorproben an Zentren weltweit mit den dort jeweils gängigen Standardverfahren erreichte der Algorithmus mehr als 99% Genauigkeit. Bereits geplante Folgestudien müssen nun zeigen, ob sich das Verfahren im klinischen Alltag bewährt und sogar weiter beschleunigen lässt.

Bildquelle: Universitätsklinikum Heidelberg
Die Methoden zur Klassifizierung von Hirntumoren haben sich rapide weiterentwickelt. Heute spielt die Molekulardiagnostik der genetischen Eigenschaften inklusive chemischer Modifikationen der Erbinformation eine zentrale Rolle. Zwar lassen sich damit seitdem Tumorarten, -subtypen und -stadien besser unterscheiden und in Folge gezielt behandeln. Die Genauigkeit bringt aber auch Probleme mit sich: Moderne Analysegeräte und -prozesse erfordern erhebliche Investitionen und die Auswertung der anfallenden Datenmenge einiges an Zeit. „Der Bedarf an umfassenden, schnellen und leichter zugänglichen Methoden ist groß“, sagt Felix Sahm, W3-Professor für Neuropathologie, stellvertretender Ärztlicher Direktor der Abteilung Neuropathologie und Leiter der Sektion Molekulare Neuropathologie am Universitätsklinikum Heidelberg (UKHD), der mit seinem Team die neuen Analyseplattformen entwickelte.
Kernstück des neuen Klassifizierungstools ist die sogenannte Nanopore-Sequenzierung: eine schnelle und auf kleinen, kostengünstigen Geräten anwendbare Methode, um die Erbinformation der Tumorzellen auszulesen und charakteristische chemische oder genetische Veränderungen zu erkennen. Die durch die Sequenzanalyse erhaltenen Daten werden unmittelbar mit den bekannten Tumorklassen abgeglichen. So könnte nach weiterer Verfeinerung der Methode künftig bereits während des operativen Eingriffs eine Tumorklassifizierung vorliegen und die Operationsstrategie angepasst werden. Die Grundlagen für das neue Diagnostik-System wurden an Universität, UKHD, DKFZ und Hopp Kindertumorzentrum (KiTZ) entwickelt und getestet. Es ist eine Kombination aus dem Schnelltest „Rapid-CNS2“, der 91 Tumorklassen unterscheidet, und „MNP-Flex“ (Heidelberg Molecular Neuropathology (MNP) methylation classifier), der die Ergebnisse unterschiedlicher Analysemethoden auswerten kann und innerhalb von 24 Stunden 184 Tumorunterklassen erkennt. „Die Kombination dieser beiden Werkzeuge deckt das gesamte Spektrum diagnostisch und therapeutisch relevanter Informationen ab“, so Prof. Dr. Dr. Felix Sahm, der auch als Wissenschaftler am DKFZ und im Deutschen Konsortium für Translationale Krebsforschung (DKTK) tätig ist.
Dieser Artikel könnte Sie auch interessieren

News • Meningeom-Forschung
Hirntumoren: bessere Klassifizierung bringt Vorteile für Diagnose und Therapie
Eine internationale Studie mit rund 3000 Patienten bestätigt die Aussagekraft eines neuen Klassifizierungssystems für Meningeome. Es kombiniert Gewebecharakteristika (Histologie) mit molekularen Analysen und verbessert so die Therapieplanung.
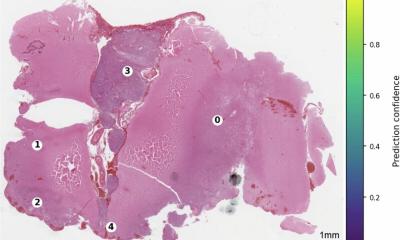
Für die präzise Klassifizierung von sehr kleinen Gewebemengen hat das Team von Prof. Sahm gemeinsam mit Forschenden des Universitätsklinikums Erlangen ein Verfahren, das bislang nur für Forschungszwecke genutzt wird, für den Einsatz in der klinischen Diagnostik weiterentwickelt. Bei der „NeuroPathology Spatial Transcriptomic Analysis“ (NePSTA) wird ein einziger Gewebeschnitt aus dem Tumor Punkt für Punkt auf jeweils mehrere tausend Krebsmarker gleichzeitig untersucht.
Besonders bei inoperablen Tumoren ist es schwierig, an eine ausreichende Menge an Tumorgewebe für die gängige Diagnostik und Klassifizierung zu kommen
Felix Sahm
Eine Künstliche Intelligenz wertet die Daten aus und kann anschließend die Ergebnisse verschiedener gängiger Analysemethoden, quasi „all-in-one“, simulieren. Das hat zwei Vorteile: Schon aus geringen Gewebemengen gewinnt dieses Verfahren eine Fülle an Informationen. Außerdem lassen sich durch die hohe räumliche Auflösung selbst kleinste Gruppen hoch aggressiver Tumorzellen erkennen, die mit gängigen Methoden nicht erfasst werden, aber für die Tumorklassifikation relevant sind.
„Besonders bei inoperablen Tumoren ist es schwierig, an eine ausreichende Menge an Tumorgewebe für die gängige Diagnostik und Klassifizierung zu kommen“, sagt Prof. Sahm. Eine mögliche Lösung bietet die sogenannte räumlich aufgelöste Transkriptom-Analyse. Dabei bezeichnet der Begriff „Transkriptom“ die Gesamtheit aller in einer Zelle für die Proteinproduktion abgelesenen Gene, die dann in einer Art „Arbeitskopie“ (mRNA) für die weitere Prozessierung vorliegen. In einer Krebszelle gibt die Transkriptom-Analyse unter anderem Auskunft darüber, ob bestimmte Gene häufiger oder seltener abgelesen werden als in gesunden Zellen oder bestimmte genetische Veränderungen vorliegen. Bei der von Prof. Sahms Team verwendeten Methode wird dazu ein wenige Mikrometer dicker Gewebeschnitt, der zuvor in Paraffin eingebettet wurde, auf einen speziellen Objektträger aufgetragen. Auf diesem befinden sich – Pixel für Pixel – Bindestellen für die „Kopien“ von jeweils mehreren tausend krebsrelevanten Genen, die, wenn eine Bindung erfolgt, ein Signal in räumlicher Auflösung erzeugen.
Das Team trainierte den Algorithmus mit den Daten von 130 Patienten mit Tumoren des Zentralen Nervensystems aus vier medizinischen Zentren. Aktuell klassifiziert NePSTA Tumoren bereits mit einer Treffsicherheit von knapp 90%. Bevor NePSTA in der Diagnostik genutzt werden kann, muss die Praxistauglichkeit noch in klinischen Studien überprüft werden.
Quelle: Universitätsklinikum Heidelberg
11.07.2025