

News • Neurodegeneration
Chorea Huntington: Neue Studie gibt Anlass zur Hoffnung
Im New England Journal of Medicine wurde jetzt eine wegweisende Studie zur Therapie der Chorea Huntington publiziert. Bei dieser erblichen, neurodegenerativen Erkrankung entwickelt sich eine progrediente Bewegungsstörung mit unkoordinierten Muskelkontraktionen. Eine wirksame Therapie gibt es derzeit nicht.
Nun konnte gezeigt werden, dass mit sogenannten Antisense-Oligonukleotiden das mutierte Gen quasi stillgelegt werden kann. Das sei ein durchaus vielversprechendes Zeichen. Ob sich das Ergebnis auch in einen bedeutsamen klinischen Nutzen übersetzt, soll nun eine internationale Multicenter-Phase 3-Studie klären, an der sich auch fünf Zentren in Deutschland beteiligen. Die Studie wurde im New England Journal of Medicine veröffentlicht.
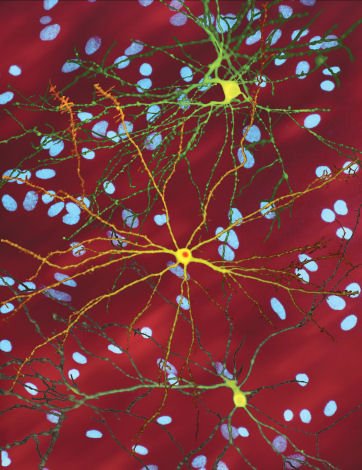
Dr. Steven Finkbeiner, Gladstone Institute of Neurological Disease, The Taube-Koret Center for Huntington's Disease Research, and the University of California San Francisco, Neuron with mHtt inclusion, CC BY 3.0
Die Chorea Huntington beginnt schleichend und wird typischerweise mit 35 bis 40 Jahren symptomatisch (möglich aber auch schon im Kindesalter). In Westeuropa sind ca. 7-10/100.000 Menschen betroffen. Der unaufhaltsame Untergang von Gehirnzellen (Neuronen) führt zu einer Bewegungsstörung, bei der ausfahrende, unkoordinierte bzw. unwillkürliche Muskelkontraktionen auftreten – bei gleichzeitig schlaffem Muskeltonus (daher der Name „Chorea“, er stammt aus dem Griechischen und bedeutet „Tanz“).
Zu Beginn stehen eine motorische Verlangsamung sowie unterschiedlich ausgeprägte psychische Veränderungen im Vordergrund. Die Erkrankung ist chronisch fortschreitend, es kommt schließlich zur völligen Pflegebedürftigkeit, Demenz und nach durchschnittlich 15 Jahren zum Tod der Patienten.
Das Huntington-Gen wird über den monogenen, autosomal-dominanten Erbgang weitergegeben, das Erkrankungsrisiko für die Kinder der Patienten beträgt 50%. Wenn das mutierte Gen vererbt wurde, kommt die Krankheit auch immer zum Ausbruch. Die ursächliche Genmutation liegt auf dem Chromosom 4; dort findet sich eine pathologisch häufige Wiederholung einer Dreibasen-Abfolge, wodurch die DNA im Zellkern nicht richtig abgelesen werden kann: Im Genprodukt, dem Huntingtin-Protein, kommt es zur fehlerhaften Molekülstruktur.
Dieser Artikel könnte Sie auch interessieren
News • Tödliche Krankheit
Ausbruch und Verlauf der Huntington-Krankheit vorhersagen
Die Ablagerungen, die bei der unheilbaren Krankheit Chorea Huntington im Gehirn der Patienten entstehen, wachsen aus kurzen Proteinfasern, berichten Forscher vom Max-Delbrück-Centrum für Molekulare Medizin in der Helmholtz-Gemeinschaft. Die Ergebnisse könnten die Diagnostik und die Suche nach neuen Medikamenten verbessern.
Für die Huntington-Erkrankung gibt es bislang keine Therapie, die den Krankheitsverlauf aufhalten kann. Das zunehmende Verständnis der komplexen molekulargenetischen Pathomechanismen hat jedoch endlich zur Entwicklung verschiedener neuer Therapieansätze geführt, von denen derzeit viele erprobt werden. „Die Therapien setzen an bestimmten Stellen des DNA-Ableseprozesses bzw. der Huntingtin-Expression, also der Bildung des fehlerhaften Proteins, an“, erläutert Frau Prof. Christine Klein, Präsidentin der Deutschen Gesellschaft für Neurologie (DGN) und u.a. auch Mitglied der DFG-Senatskommission für Grundsatzfragen in der Klinischen Forschung. Beim DNA-Ableseprozess wird im Zellkern immer zunächst messenger-RNA (mRNA) gebildet, die sozusagen als Matrize für die Proteinsynthese dient. Bei dem Therapieansatz mit sogenannten Antisense-Oligonukleotiden (ASOs) wird diese Matritze und somit die Bildung von Htt-Protein durch synthetisch hergestellte spiegelbildliche (komplementäre) mRNA-Bausteine (Antisense-Oligonukleotide) spezifisch gehemmt. Das mutante Htt-Gen wird praktisch stillgelegt („gene silencing“). Da ASOs die Blut-Hirn-Schranke nicht passieren können, müssen sie intrathekal (d. h. in den in den Liquorraum) injiziert werden.
Es ist durchaus ein vielversprechendes Zeichen, dass die Expression des fehlerhaften Huntingtin-Proteins im Liquor reduziert werden konnte
Alexander Münchau
An der neuen Phase-1-2a-Studie mit dem Oligonukleotid HTTRx waren weltweit Zentren in Kanada, U.K. und Deutschland (Berlin, Bochum, Ulm) beteiligt. 46 erwachsene Patienten in einem frühen Erkrankungsstadium wurden 3:1 doppelblind randomisiert. 34 Patienten erhielten viermal, jeweils im Abstand von vier Wochen verschiedene Dosierungen HTTRx intrathekal (10, 30, 60, 90 oder 120 mg), zwölf erhielten Placebo. Primärer Endpunkt war die Sicherheit der Substanz, sekundär wurde im Liquor die Pharmakokinetik von HTTRx sowie die Konzentrationen von Huntingtin ermittelt. Die Substanz wurde insgesamt gut vertragen, alle Patienten durchliefen die Studie vollständig ohne Studienabbrüche. Ernste Nebenwirkungen traten nicht auf.
Im Ergebnis führte die Behandlung mit HTTRx dosisabhängig zur Absenkung der Htt-Konzentrationen im Liquor: Während in der Placebogruppe die Konzentration um 10% zunahm, sank sie in den HTTRx-Gruppen mit steigender HTTRx-Dosierung um -20%, -25%, -28%, -42% und -38% ab. Funktionelle neurologische, kognitive und psychiatrische Tests zeigten keine Unterschiede vom Studienbeginn bis zum Ende; auch nicht zwischen Placebo- und den Verumgruppen. Eine (nicht-präspezifizierte) Posthoc-Analyse verglich den Verlauf des cUHDRS-Scores (“composite Unified Huntington Disease Rating Scale”) als Maß der Krankheitsprogression mit den Htt-Liquorkonzentrationen: In zwei von vier Teilergebnissen gab es dabei positive Korrelationen zwischen sinkenden Htt-Werten und einer Verbesserung des cUHDRS-Scores. Die Autoren weisen jedoch ausdrücklich darauf hin, dass die Studie statistisch nicht für diese Analyse konzipiert war und daher keine validen Aussagen zu einer klinischen Wirksamkeit hinsichtlich einer Progressionshemmung gemacht werden können.
„Es ist durchaus ein vielversprechendes Zeichen, dass die Expression des fehlerhaften Huntingtin-Proteins im Liquor reduziert werden konnte. Noch wissen wir aber nicht abschließend, in wie weit die Htt-Konzentration krankheitsauslösend ist oder nur einen Surrogatparameter darstellt“, erklärt Prof. Dr. med. Alexander Münchau, Leiter des Lübecker Zentrums für Seltene Erkrankungen (ZSE), einer fachübergreifenden Einrichtung der Universität zu Lübeck und des Universitätsklinikums Schleswig-Holstein - Campus Lübeck. „Es muss betont werden, dass bislang der sichere Beleg dafür fehlt, dass es durch die Senkung von Huntingtin im Liquor auch zu einer Verbesserung des klinischen Bildes kommt. Die Patientenzahl und die Studiendauer waren in dieser Phase-2-Studie dafür einfach noch nicht ausreichend.“
Ende vergangenen Jahres begann weltweit (in 15 Ländern, 46 Standorte, davon fünf deutsche Zentren) die Phase-3-Studie „GENERATION-HD1“, die bis 2022 laufen soll. Insgesamt sollen 660 symptomatische Patienten eingeschlossen werden und verschiedene Dosen RG6042 (RO7234292) gegen Placebo getestet werden. „Das Studienprotokoll sieht neben umfangreichen Laboruntersuchungen auch eine Reihe klinischer Tests zum motorischen, kognitiven und psychosozialen Krankheitsverlauf vor“, erklärt Prof. Münchau, „so dass wir hoffentlich in absehbarer Zeit wissen, ob die Therapie auch die dringend erhofften klinischen Wirkungen zeigt.“
Quelle: Deutsche Gesellschaft für Neurologie
08.05.2019

{kind=link}