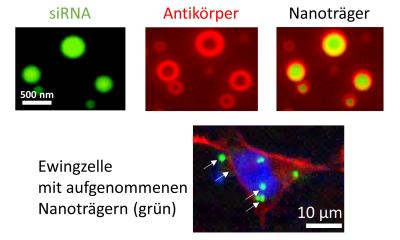
Quelle: F. Cidre Aranaz/ KiTZ
News • Knochen- und Weichteilkrebs
Einblicke in die Metastasenbildung bei Ewing-Sarkomen
Ewing-Sarkome sind hochaggressive Tumoren, die sich in Knochen- oder Weichteilgeweben bilden können und hauptsächlich bei Kindern und Jugendlichen vorkommen. Bei der Ausbreitung von Metastasen spielen regulatorische Proteine eine Schlüsselrolle, die hierfür bestimmte genetische Netzwerke aktivieren. Einige dieser Schlüsselproteine könnten für die Vorhersage des Krankheitsverlaufes von Patienten genutzt werden, wie eine aktuelle Studie zeigt.
Nach derzeitigen Standardtherapien kommt es bei etwa einem Drittel der Ewing-Sarkom-Patienten zu Rückfällen – oft mit fatalem Ausgang. Um zu verstehen, wie diese Krebserkrankung weiter fortschreitet und Metastasen entstehen, hat ein Wissenschaftlerteam des Hopp-Kindertumorzentrums Heidelberg, des Deutschen Krebsforschungszentrums (DKFZ) und der Ludwigs-Maximilians-Universität München im Rahmen eines von der Wilhelm Sander-Stiftung geförderten Forschungsprojektes die molekularen Treiber, die zu einem Rückfall und zur Metastasenbildung beim Ewing-Sarkom führen können, untersucht.
Eine wichtige Rolle spielt dabei ein genetischer Defekt, der zu einer krebstreibenden Mutation im Gen EWSR1-ETS führt. Das vom mutierten EWSR1-ETS abgelesene Protein aktiviert bestimmte genregulatorische Netzwerke, die über den Krankheitsverlauf entscheiden. Wie die vorliegende Studie an Ewing-Sarkom-Zellen zeigt, reguliert das mutierte Schlüsselprotein in den Tumorzellen mehrere hundert Gene, die in komplexen Netzwerken miteinander interagieren.
Die Forscher korrelierten diese Daten mit dem Krankheitsverlauf von 166 Ewing-Sarkom-Patienten und identifizierten auf diese Weise einen besonders vielversprechenden Schalter in diesen genetischen Netzwerken, der als Biomarker dienen könnte: Bei Patienten mit ungünstigem Krankheitsverlauf produzierten die Tumorzellen nur wenige Mengen des genregulatorischen Proteins TCF7L1. Zudem fanden die Wissenschaftler in Metastasen deutlich weniger TCF7L1 als noch in den Primärtumoren. „Wir gehen deshalb davon aus, dass die geringen Mengen dieses Proteins dazu führen, dass genetische Netzwerke aktiviert werden, die den aggressiven Krankheitsverlauf und die Bildung von Metastasen eher begünstigen“, erklärt Prof. Dr. Dr. Thomas Grünewald, Arbeitsgruppenleiter am KiTZ, Abteilungsleiter am DKFZ und dem Universitätsklinikum Heidelberg (UKHD).
Umgekehrt konnte eine wiederhergestellte TCF7L1-Produktion die Ausbreitung von Metastasen unterdrücken, so zeigten Untersuchungen in Mäusen. Künftig könnte TCF7L1 als prognostischer Biomarker beim Ewing-Sarkom geprüft werden, vermutet auch Dr. Florencia Cidre-Aranaz, die mit Thomas Grünewald die Studie leitete: „Die Menge an TCF7L1 in den Tumorproben ließe sich z. B. durch Anfärben in Gewebeschnitten bestimmen“, sagt Cidre-Aranaz vom KiTZ und vom DKFZ.
Dieser diagnostische Ansatz ließe sich möglicherweise auch auf andere Krebsarten, wie Brustkrebs, Darmkrebs oder akute lymphatische Leukämie übertragen, in denen die Fehlregulierung von TCF7L1 ebenfalls das Tumorwachstum zu begünstigen scheint. Erste Ergebnisse aus dem Labor deuten zudem darauf hin, dass TCF7L1 möglicherweise auch eine geeignete Zielstruktur sein könnte, um Ewing-Sarkome bei Kindern und Jugendlichen zu behandeln und eine Metastasierung zu verhindern. Diesem Ansatz wollen die Forscher in Folgestudien weiter nachgehen.
Die Ergebnisse des Forschungsteams wurden in der Fachzeitschrift Molecular Cancer veröffentlicht.
Quelle: Wilhelm Sander-Stiftung
07.01.2022