Bildquelle: Adobe Stock/alphaspirit
News • 'Synthetischer Ursprung' von SARS-CoV-2
Kommt das Coronavirus aus dem Labor? Experten zweifeln neue Analyse an
In einem auf BioRxiv publizierten Preprint legen die Autoren statistische Analysen der Genomsequenz von SARS-CoV-2 vor, aus denen sie folgern, dass dieses Virus mit hoher Wahrscheinlichkeit synthetisch entstanden ist. Experten der Universität Würzburg haben das Preprint begutachtet.
In der Summe ergibt sich aus den in der Studie vorgelegten Analysen keine sichere Evidenz für die von den Autoren formulierte Schlussfolgerung, dass SARS-CoV-2 synthetischen Ursprungs sei. Die Frage nach dem genauen Ursprung des Virus, das die aktuelle Covid-19-Pandemie ausgelöst hat, bleibt damit weiterhin offen.
In dem auf BioRxiv publizierten, bisher nicht durch Experten begutachteten Preprint präsentieren die Autoren Analysen, die nach ihrer Interpretation eine „synthetische Entstehung“ von SARS-CoV-2 und dessen Freisetzung im Rahmen eines „Laborunfalls“ nahelegen.
Kernaussage des Preprints ist, dass das Genom von SARS-CoV-2 ein „auffälliges Muster“ an Schnittstellen für bestimmte Restriktionsenzyme (BsaI und BsmBI) aufweist und daher mit hoher Wahrscheinlichkeit nicht durch natürliche Evolution entstanden ist. Die Autoren kommen aufgrund statistischer Analysen zu dem Schluss, dass dieses Schnittstellenmuster höchstwahrscheinlich im Zuge der Etablierung eines Reversen Genetik Verfahrens für das – wie man annimmt – aus Fledermäusen stammende SARS-CoV-2-Ursprungsvirus in einem Forschungslabor als „Fingerabdruck“ im SARS-CoV-2 Genom entstanden ist.
Restriktionsenzyme wie BsaI und BsmBI werden unter anderem eingesetzt, um Genome von aus Tieren wie Fledermäusen isolierte Coronaviren zu klonieren und somit erforschbar zu machen. Solche sogenannten Reverse-Genetik-Verfahren sind für die virologische Forschung von großer Bedeutung. Sie erlauben die genetische Konservierung ansonsten mutationsanfälliger Viren sowie die Erforschung einzelner viraler Genfunktionen. Die mit knapp 30.000 Nukleotiden relativ großen Coronavirusgenome können so in 5-8 kleinen Teilstücken in Bakterien kloniert und verändert werden. Für die Klonierung einzelner Teilstücke müssen gelegentlich einzelne Basen des viralen Genoms ausgetauscht werden, um die für die Klonierung notwendigen Schnittstellen einzufügen bzw. unerwünschte Schnittstellen zu entfernen. Anschließend lassen sich die einzelnen Teilstücke wieder zu einem kompletten Virusgenom zusammenfügen.
Die Würzburger Experten Florian Erhard, Oliver Kurzai und Lars Dölken aus dem Institut für Virologie und Immunbiologie und dem Institut für Hygiene und Mikrobiologie der Universität Würzburg, haben das Preprint einer wissenschaftlichen Beurteilung unterzogen. Ihre Kernaussagen lauten:
1. Anders als von den Autoren behauptet, kann das Schnittstellenmuster auch natürlich entstanden sein – ähnliche Muster finden sich auch bei eng mit SARS-CoV-2 verwandten Coronaviren
In die Analysen im Preprint wurden offensichtlich einige bekannte, nah mit SARS-CoV-2 verwandte Viren nicht mit in die durchgeführten Analysen aufgenommen
Florian Erhard, Oliver Kurzai, Lars Dölken
Alle 5 Schnittstellen (BsmBI (n=3) und BsaI (n=2)), die für die Analysen im Preprint von zentraler Bedeutung sind, finden sich auch häufig in nahe verwandten Coronaviren. Die Existenz dieser 5 Schnittstellen im SARS-CoV-2-Genom lässt sich also auch ohne menschliche Manipulation erklären. In die Analysen im Preprint wurden offensichtlich einige bekannte, nah mit SARS-CoV-2 verwandte Viren nicht mit in die durchgeführten Analysen aufgenommen.
Die Autoren des Preprints argumentieren weiterhin, dass für genetische Arbeiten ungünstige Schnittstellen im SARS-CoV-2-Genom fehlen und mutmaßlich künstlich verändert („gelöscht“) wurden. Während viele andere Coronaviren tatsächlich deutlich mehr Schnittstellen für die beiden analysierten Restriktionsenzyme aufweisen, zeigen einige nahverwandte Fledermaus-Coronaviren wie BANAL-20-103 und BANAL-116 ebenfalls nur 5 bzw. 7 Schnittstellen bei ähnlich großen Genomfragmenten.
2. Die Position der analysierten Restriktionsendonuklease-Schnittstellen im Bereich des S-Gens ist kein sicherer Hinweis auf eine gentechnische Manipulation des SARS-CoV-2 Genoms
Das Spike-Protein von Coronaviren ist von besonderem Interesse, da es ganz wesentlich bestimmt, ob menschliche Zellen infiziert werden können oder nicht. Für Reverse-Genetik-Modelle war daher von besonderem Interesse, den für das Spike-Protein kodierenden Bereich des Coronavirusgenoms mit geringem Arbeitsaufwand austauschen bzw. verändern zu können. Wie die Autoren zeigen, ließe sich mit Hilfe der beiden Schnittstellen für BsaI der wichtigste Teil des Spike-Proteins von SARS-CoV-2, nämlich die Rezeptor-bindende Domäne (RBD) sowie die Furin-Schnittstelle, leicht manipulieren. Für kein anderes Coronavirusisolat erscheint dies so einfach mit BsaI möglich zu sein, da entweder die entsprechenden Schnittstellen fehlen bzw. zusätzlich Schnittstellen beim Verdau mit BsaI zu einer Zerstückelung des jeweiligen Genoms führen würden.
Dieser Artikel könnte Sie auch interessieren

News • Mechanismus entschlüsselt
Forscher klären Aktivierung von SARS-CoV-2
Das SARS-Coronavirus 2 (SARS-CoV-2) infiziert Lungenzellen und ist für die Covid-19-Pandemie verantwortlich. Das sogenannte Spike-Protein dient dem Virus als Schlüssel zur Wirtszelle und trägt eine ungewöhnliche Aktivierungssequenz.
Die Autoren argumentieren, dies spreche dafür, dass das SARS-CoV-2 Genom für den Austausch und die Manipulation der wichtigen Rezeptor-bindenden Domäne sowie der Furin Schnittstelle des Spike Proteins optimiert wurde. Tatsächlich wurde die Kombination von BsaI und BsmBI in der Vergangenheit von Arbeitsgruppen in Wuhan eingesetzt, um Coronavirusgenome aus Fledermäusen zu klonieren und sogenannte Gain-of-Function-Experimente durchzuführen. Allerdings wurden hierbei die beiden BsaI-Schnittstellen jeweils so positioniert, dass sie den Austausch des gesamten Spike-Proteins ermöglichten. Dies ist für SARS-CoV-2 so nicht möglich. Hätten sich die beiden BsaI-Schnittstellen an exakt der gleichen Stelle befunden wie in bereits veröffentlichten Reverse-Genetik-Modellen, so wäre dies in der Tat ein starker Hinweis auf einen Laborunfall gewesen. So bleibt allerdings festzustellen, dass genau diese beiden BsaI-Schnittstellen häufig auch bei nahe verwandten Coronaviren von SARS-CoV-2 vorkommen. Allerdings besitzen Coronaviren in aller Regel mindestens eine weitere BsaI-Site, die also ausgeschaltet, d.h. mutiert, hätte werden müssen. Anbetracht der hohen Mutationsrate zirkulierender SARS-CoV-2-Varianten sollte man zudem annehmen, dass künstlich eingefügte synonyme (wobble) Mutationen, die zur Erzeugung bzw. Elimination von Restriktionsschnittstellen eingefügt wurden, im Laufe der mehr als zweijährigen Pandemie wieder verschwinden und künstlich eliminierte wieder erscheinen. Tatsächlich haben aber die Omikron-Varianten dasselbe Schnittstellen-Verteilungsmuster wie das Originalvirus aus Wuhan.
3. Die statistischen Analysen der Arbeit zur Verteilung von Restriktionsenzym-Schnittstellen sind in wesentlichen Punkten fehlerhaft bzw. unvollständig
Die Analyse einer einzigen, selektiv ausgewählten Kombination von 2 Restriktionsenzyme (hier: BsaI und BsmBI) ist nicht geeignet um einen molekularbiologischen Eingriff zu beweisen.
Florian Erhard, Oliver Kurzai, Lars Dölken
Die im Preprint analysierte Kombination von BsaI und BsmBI eignet sich für die allermeisten Coronaviren tatsächlich nicht, um deren Genom in eine geeignete Anzahl (5 bis 7) von Fragmenten geeigneter Größe (<8000 Nukleotide) zu zerlegen (Fig. 3C im Preprint; siehe unten). Wie eigene Analysen von uns aber zeigten, ist dies unter Einbeziehung anderer, ähnlicher Restriktionsenzyme problemlos möglich. Unter Nutzung der im Preprint verwendeten und nachvollziehbar dokumentierten Algorithmen kann man zeigen, dass sich praktisch für jedes Coronavirusgenom eine entsprechend geeignete Kombination von Restriktionsenzymen finden lässt. Die Analyse einer einzigen, selektiv ausgewählten Kombination von 2 Restriktionsenzyme (hier: BsaI und BsmBI) ist nicht geeignet um einen molekularbiologischen Eingriff zu beweisen. Analysiert man lediglich eine, für ein bestimmtes Virus passende Kombination von zwei Restriktionsenzymen, ist es naheliegend, dass diese Kombination bei anderen Virusisolaten signifikant schlechter geeignet ist, um ein Reverses-Genetik-Modell zu konstruieren. Dies führt zu der Fehlinterpretation, dass das Virus, in diesem Fall SARS-CoV-2, für das die Auswahl der analysierten Kombination von Restriktionsendonukleasen optimiert wurde, nicht natürlichen Ursprungs ist.
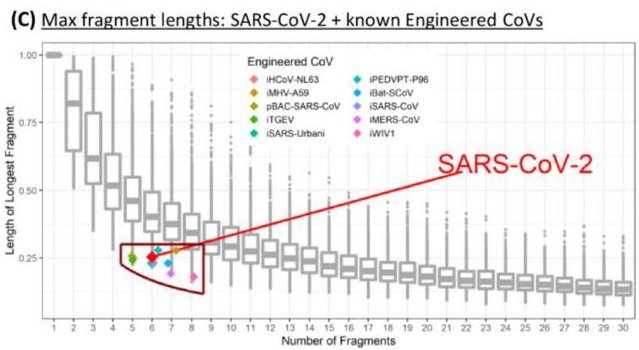
Bildquelle: Bruttel et al., bioRxiv preprint 2022 (CC BY-NC-ND 4.0)
4. Die Analysen zur in-silico-Evolution von zwei nahverwandten Coronaviren mit dem Ziel, ein vergleichbares Schnittmuster wie bei SARS-CoV-2 zu bekommen (Fig. 4 im Preprint), sind nicht überzeugend
Die Annahme rein zufälliger Mutationen in einem Virusgenom ist nicht zulässig, da die meisten Mutationen die Aminosäuresequenz der viralen Proteine stören bzw. zerstören. Zudem hätten die Wissenschaftler auch hier ebenfalls mit allen akzeptablen Kombinationen von Restriktionsenzymen arbeiten müssen.
Die im Preprint beschriebene Berechnung der Wahrscheinlichkeit, mit der das beobachtete Schnittmuster von SARS-CoV-2 natürlich entstanden sein kann, ist fehlerhaft. Die Autoren kombinieren hierzu die Wahrscheinlichkeiten für insgesamt 5 verschiedene Kriterien. Diese sind jedoch weder voneinander unabhängig, noch ist die verwendete Methode zur Kombination dieser Wahrscheinlichkeitswerte geeignet. Zudem ist jede einzelne verwendete Wahrscheinlichkeit von den gleichen potentiellen Fehlerquellen, wie oben aufgeführt, betroffen.
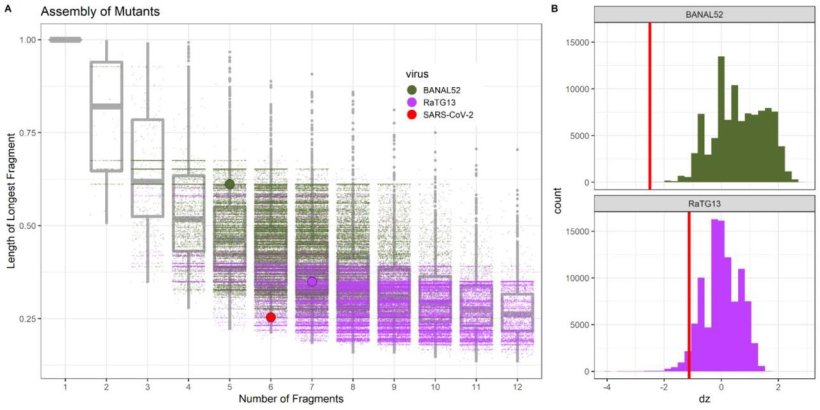
Bildquelle: Bruttel et al., bioRxiv preprint 2022 (CC BY-NC-ND 4.0)
Zusammenfassung
In dem begutachteten Preprint legen die Autoren statistische Analysen der Genomsequenz von SARS-CoV-2 vor, aus denen sie folgern, dass dieses Virus mit hoher Wahrscheinlichkeit synthetisch entstanden ist. Der Preprint ist sorgsam ausgearbeitet und erfüllt die grundlegenden wissenschaftlichen Anforderungen insbesondere im Hinblick auf eine einwandfreie und transparente Darstellung der verwendeten Methodik. Die im Preprint dargestellten Analysen weisen jedoch erhebliche methodische Schwachstellen auf. Diese führen dazu, dass wesentliche Schlussfolgerungen der Autoren einer wissenschaftlichen Überprüfung nicht standhalten bzw. überinterpretiert wurden. Im Gegensatz zu den im Preprint formulierten Aussagen ist das im Genom von SARS-CoV-2 gefundene Muster an Restriktionsenzym-Schnittstellen nicht mit hinreichend hoher Wahrscheinlichkeit durch genetische Manipulation zu erklären. In der Summe ergibt sich aus den in der Studie vorgelegten Analysen keine sichere Evidenz für die von den Autoren formulierte Schlussfolgerung, dass SARS-CoV-2 synthetischen Ursprungs sei. Die Frage nach dem genauen Ursprung von SARS-CoV-2 bleibt damit weiterhin offen.
Quelle: Universitätsklinik Würzburg
27.10.2022