© HHU / Sara Schulte
News • Bioinformatik
HOGVAX: ein neuer Ansatz zur Impfstoffentwicklung
Impfstoffe sollen möglichst viele Menschen vor Infektionen schützen. Kurze Proteinfragmente des Erregers, sogenannte Epitope, gelten als ein vielversprechender neuer Ansatz für die Impfstoffentwicklung.
Bioinformatiker der Heinrich-Heine-Universität Düsseldorf (HHU) stellen nun in der Fachzeitschrift Cell Systems ein Verfahren vor, um diejenigen Epitope zu identifizieren, die bei einer möglichst breiten Bevölkerungsgruppe eine sichere Impfreaktion versprechen. Sie haben mit ihrem Tool HOGVAX auch Impfstoffkandidaten gegen das Coronavirus Sars-CoV-2 berechnet.
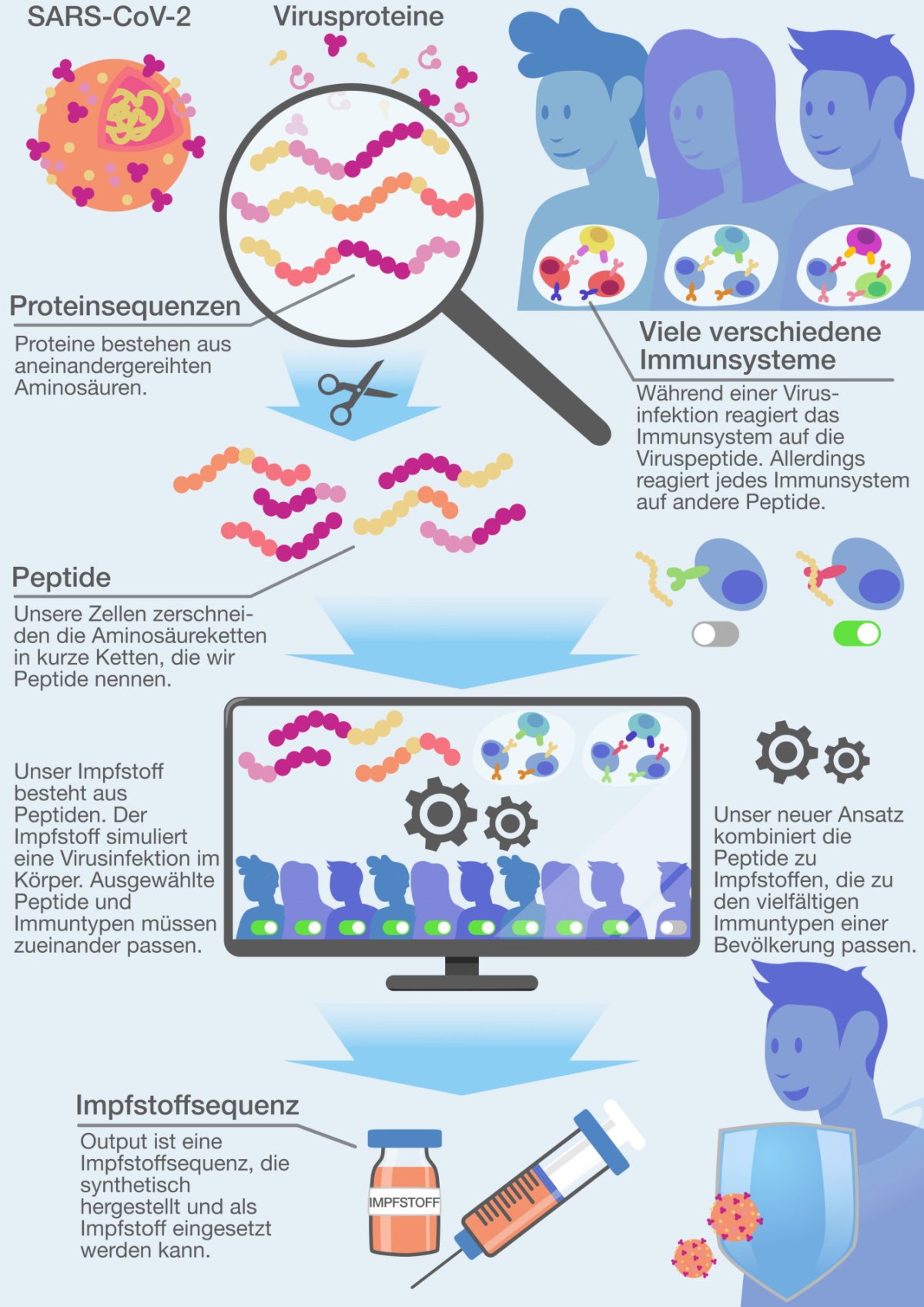
Während der Corona-Pandemie haben sich insbesondere sogenannte mRNA-Impfstoffe als erfolgreich und flexibel erwiesen. Diese Impfstoffe zielen auf die sogenannten Spikeproteine – charakteristische Strukturen auf der Oberfläche des Virus. Die mRNA enthält die Sequenz des Spikeproteins, das nach der Impfung im Körper hergestellt wird und dann das menschliche Immunsystem trainiert. Als alternative Methode zur mRNA werden „Epitope“ angesehen – kurze Fragmente von Proteinen des Krankheitserregers, die in der Lage sind, eine Immunantwort auszulösen. Sie gelten als vielversprechend, um schnell, kostengünstig und sicher gezielte Immunantworten zu erhalten.
Jeder Mensch besitzt ein individuelles Immunsystem: Je nach Infektionsgeschichte ist die Immunabwehr auf unterschiedliche Eiweißstoffe trainiert und reagiert auf diese. „Dies ist ein grundlegendes Problem von Impfstoffen, die auf Epitopen basieren“, erläutert Prof. Dr. Gunnar Klau, Inhaber des Lehrstuhls für Algorithmische Bioinformatik der HHU. Zusammen mit seiner Doktorandin Sara Schulte und Prof. Dr. Alexander Dilthey vom Institut für Medizinische Mikrobiologie und Krankenhaushygiene hat er sich mit einem neuen Ansatz für die Entwicklung solcher Impfstoffe beschäftigt.
© HHU / Sara Schulte
Prof. Klau vergleicht die Problematik mit einem Koch, der ein neues Gericht für eine große Festveranstaltung kreieren muss: „Manche Gäste haben Allergien, andere mögen bestimmte Zutaten nicht. Der Koch will nun Zutaten auswählen, so dass möglichst viele Gäste mitessen können und es ihnen auch schmeckt.“ Übersetzt auf Impfstoffentwicklung heißt dies: Gesucht sind diejenigen Epitope, die bei möglichst vielen Menschen eine gute Immunreaktion auslösen. Denn man kann nicht beliebig viele der Proteinfragmente in einen Impfstoff packen, damit sich die verschiedenen Immunsysteme die für sie passenden Sequenzen aussuchen; dafür reicht die Kapazität des Trägermediums nicht aus.
Das Team der drei Forschenden wählt mit ihrem bioinformatischen Werkzeug „HOGVAX“ einen besonderen Ansatz. Sara Schulte: „Statt die Epitope für den Impfstoff aneinanderzuhängen nutzen wir identische Sequenzen am Anfang und Ende der Epitope aus, um diese quasi ineinander zu schieben. Der identische Teil, oder ‚overlap‘, wird also nur einmal im Impfstoff repräsentiert. Dadurch sparen wir viel Platz ein.“ Auf diese Weise können viel mehr Epitope in einen Impfstoff gepackt werden.
Um die Epitope und deren längste Überlappungen effizient zu verwalten, setzen die Forschenden eine Datenstruktur namens „hierarchical overlap graph“ (kurz HOG) ein. Klau: „Ums im Bild des Kochens zu bleiben: HOG entspricht einem komprimierten oder eben geschrumpften Kochbuch, aus dem der Koch nun die Rezepte auswählen kann, die für alle Gäste passen.“ Prof. Dilthey: „Wir haben zum Test HOGVAX auf Daten des Sars-CoV-2-Virus angewandt. Dabei konnten wir deutlich mehr Epitope integrieren als andere Tools. Hiermit wären laut unseren Berechnungen über 98% der Weltbevölkerung zu erreichen und damit immunisierbar.“
Zu den weiteren Perspektiven ihrer Ergebnisse sagt Sara Schulte: „In Zukunft werden wir daran arbeiten, HOGVAX für den Einsatz in der Krebstherapie abzuwandeln. Hier gilt es, spezifisch für den einzelnen Patienten designte Wirkstoffe zu entwickeln, die gezielt Tumorzellen angreifen.“
Quelle: Heinrich-Heine-Universität Düsseldorf
27.12.2023