News • "Enhance that"
New algorithm sharpens focus of cryo-electron microscopes
Scientists have developed a tool that improves the resolution and accuracy of powerful microscopes that are used to reveal insights into biology and medicine.
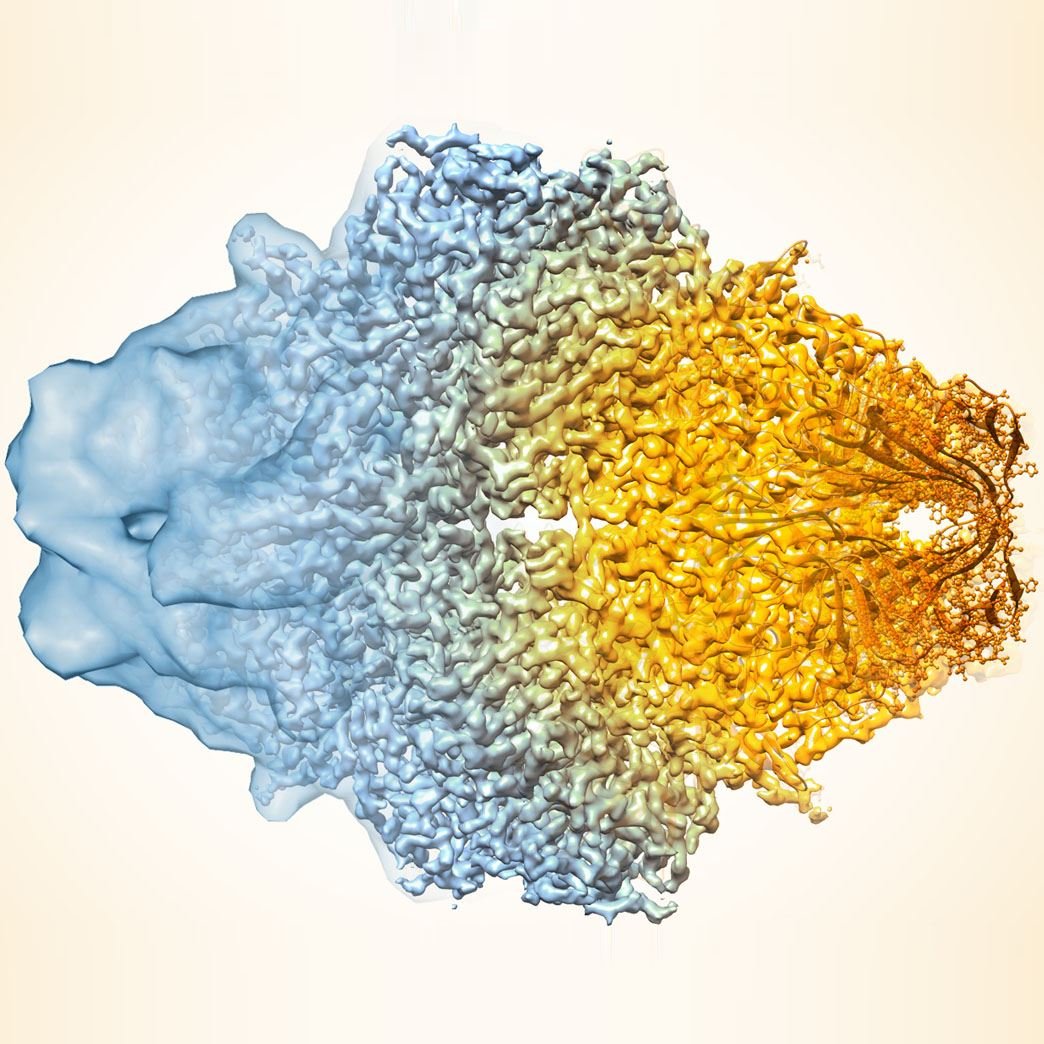
Image credit: Veronica Falconieri/National Cancer Institute
In a study published in Nature Methods, a multi-institutional team led by Tom Terwilliger from the New Mexico Consortium and including researchers from Lawrence Berkeley National Laboratory (Berkeley Lab) demonstrates how a new computer algorithm improves the quality of the 3D molecular structure maps generated with cryo-electron microscopy (cryo-EM).
For decades, these cryo-EM maps – generated by taking many microscopy images and applying image-processing software – have been a crucial tool for researchers seeking to learn how the molecules within animals, plants, microbes, and viruses function. And in recent years, cryo-EM technology has advanced to the point that it can produce structures with atomic-level resolution for many types of molecules. Yet in some situations, even the most sophisticated cryo-EM methods still generate maps with lower resolution and greater uncertainty than required to tease out the details of complex chemical reactions.
“In biology, we gain so much by knowing a molecule’s structure,” said study co-author Paul Adams, Director of the Molecular Biophysics & Integrated Bioimaging Division at Berkeley Lab. “The improvements we see with this algorithm will make it easier for researchers to determine atomistic structural models from electron cryo-microscopy data. This is particularly consequential for modeling very important biological molecules, such as those involved in transcribing and translating the genetic code, which are often only seen in lower-resolution maps due to their large and complex multi-unit structures.”
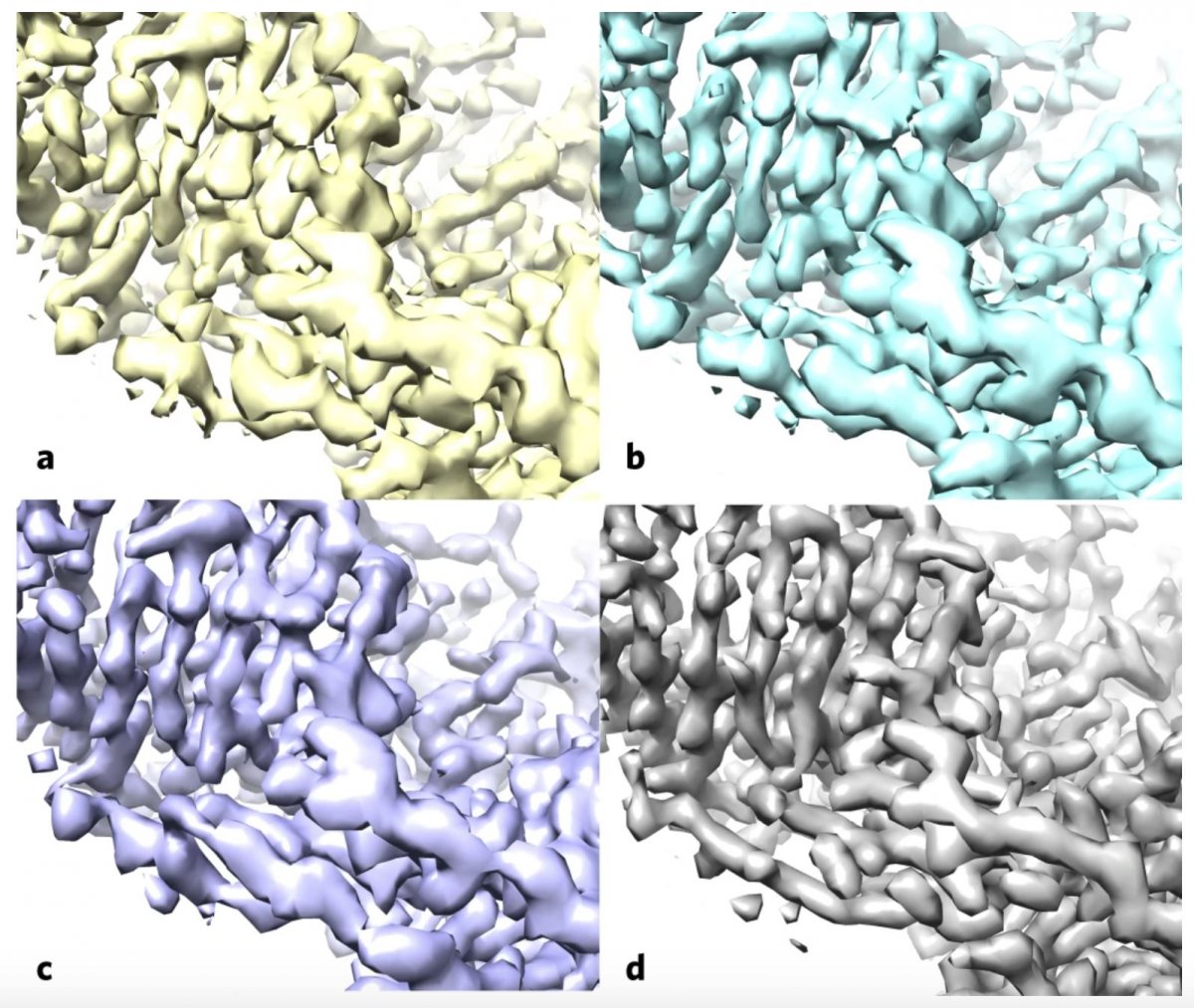
Image cCredit: Terwilliger et al./Nature Methods
The algorithm sharpens molecular maps by filtering the data based on existing knowledge of what molecules look like and how to best estimate and remove noise (unwanted and irrelevant data) in microscopy data. An approach with the same theoretical basis was previously used to improve structure maps generated from X-ray crystallography, and scientists have proposed its use in cryo-EM before. But, according to Adams, no one had been able to show definitive evidence that it worked for cryo-EM until now.
The team – composed of scientists from New Mexico Consortium, Los Alamos National Laboratory, Baylor College of Medicine, Cambridge University, and Berkeley Lab – first applied the algorithm to a publicly available map of the human protein apoferritin that is known to have 3.1-angstrom resolution (an angstrom is equal to a 10-billionth of a meter; for reference, the diameter of a carbon atom is estimated to be 2 angstroms). Then, they compared their enhanced version to another publicly available apoferritin reference map with 1.8-angstrom resolution, and found improved correlation between the two. Next, the team used their approach on 104 map datasets from the Electron Microscopy Data Bank. For a large proportion of these map sets, the algorithm improved the correlation between the experimental map and the known atomic structure, and increased the visibility of details.
The authors note that the clear benefits of the algorithm in revealing important details in the data, combined with its ease of use – it is an automated analysis that can be performed on a laptop processor – will likely make it part of a standard part of the cryo-EM workflow moving forward. In fact, Adams has already added the algorithm’s source code to the Phenix software suite, a popular package for automated macromolecular structure solution for which he leads the development team.
Source: Lawrence Berkeley National Laboratory
09.10.2020