Image credit: Dr Patricia Yeyati (University of Edinburgh)
News • Ciliopathy reseach
Rare lung disease: genetics reveals cause of PCD
Using patient cells, organoid and mouse models, researchers reveal how cilia are key to how different mutations in a single tubulin gene can result in multiple rare diseases.
An international consortium of researchers, led by the Mill group at the Institute of Genetics and Cancer and the Perrault group at the Imagine Institute in Paris, FR, uncovered new insights into primary ciliary dyskinesia (PCD), a rare disease affecting 1 in 7,500 people. The findings have now been published in Science.
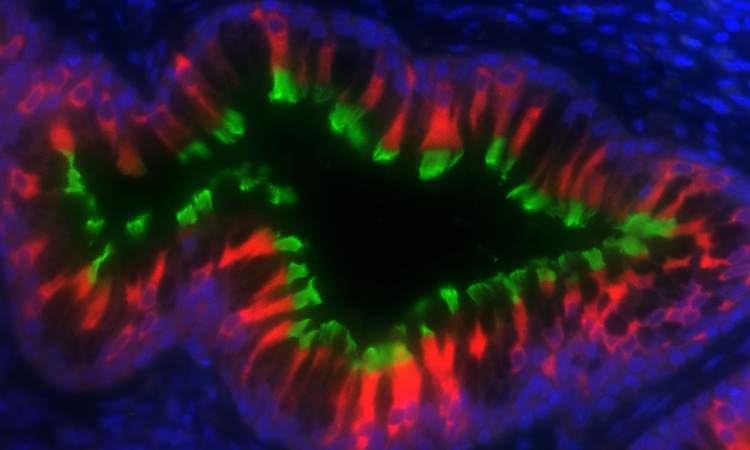
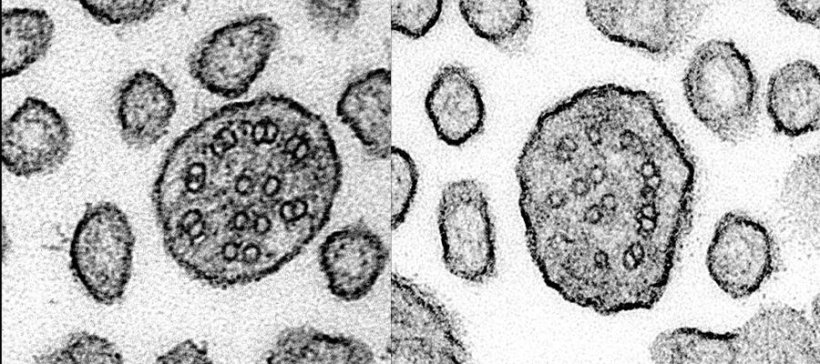
Cilia are small, hair-like structures found on the surface of most of our cells. PCD is caused by genetic mutations that specifically affect the presence and function of motile cilia, which move to create fluid flow across the surface of our airways, brain ventricles and reproductive tracts. PCD patients experience life-long, progressive decline in lung function, with other features that vary by patient and mutation, including abnormal organ formation, hydrocephaly, and infertility.
These exciting and unexpected insights into ciliopathies were only possible due to the collaborative partnership between the patients in our studies, their clinicians and researchers with interdisciplinary expertise from across the world
Pleasantine Mill
Mutations in over 50 genes account for 70% of PCD cases, with the remainder of cases unexplained. Using next-generation DNA sequencing approaches, researchers were surprised to find recurrent mutations in the beta-tubulin gene TUBB4B, in twelve different PCD patients, that were not found in their parents. Moreover, where on the TUBB4B protein these recurrent mutations were found resulted in very different clinical presentations.
TUBB4B is one of the building blocks of microtubules – scaffold-like structures involved in cell shape, movement, and cell division and critical for cilia formation. Each link of the tubulin ‘chain’ is composed of one alpha and one beta tubulin protein. Humans have 10 types of beta tubulin, including TUBB4B. The different tubulin subunits seem to not be interchangeable and may have different roles as mutations in one, such as TUBB4B, cannot be always compensated for by the others.
Researchers used both computational and laboratory-based approaches to resolve how mutations in different parts of the TUBB4B molecule differently affect how the tubulin building blocks assemble and disrupt cilia form or function. They found mutations at different interfaces between tubulin links result in different patient outcomes, such as early vision loss, lung disease, or both conditions simultaneously. The researchers were able to show that one mutant copy of TUBB4B was capable of over-riding the healthy copy to disrupt microtubule and cilia formation, a phenomenon called the ‘dominant negative’ effect, previously not observed in PCD.
In addition to improving the diagnosis and genetic counselling of PCD patients and their families, this novel disease mechanism will require different therapeutic strategies to treat these patients. "These exciting and unexpected insights into ciliopathies were only possible due to the collaborative partnership between the patients in our studies, their clinicians and researchers with interdisciplinary expertise from across the world," said Professor Pleasantine Mill, Chair of Cilia Biology at the University of Edinburgh, who co-led the study. Study authors Mill, Shoemark and Mitchison are co-leads, with other UK researchers, for a newly funded £9.4M UK-wide LifeArc Centre for Rare Respiratory Diseases, which aims to improve molecular diagnoses as well accelerate molecular insights to develop new therapies for rare lung diseases, like PCD.
"We know firsthand the unmet need from significant burden of disease for rare lung diseases like PCD. We hope this work and our newly funded collaborative centre will provide the foundation to raise awareness, develop much-needed treatments and use them to benefit these patients," added Prof. Amelia Shoemark.
Source: University of Edinburgh
29.04.2024