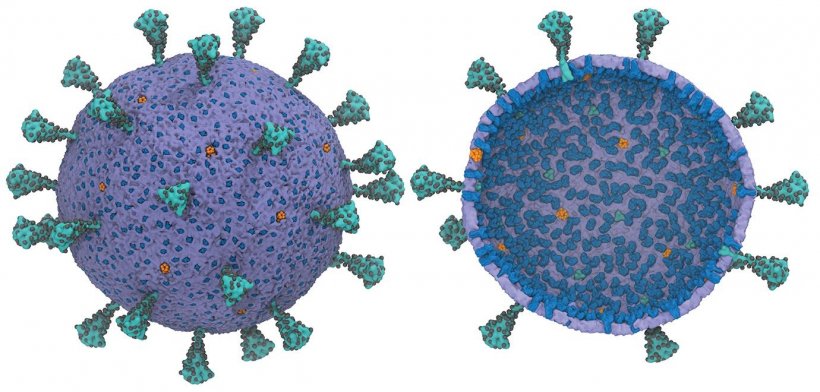
Image credit: Yu et al., “A Multiscale Coarse-Grained Model of the SARS-CoV-2 Virion,” Biophysical Journal (2021)
News • Virion recreated
First computational model of entire SARS-CoV-2 virus
Researchers at the University of Chicago have created the first usable computational model of the entire virus responsible for Covid-19—and they are making this model widely available to help advance research during the pandemic.
“If you can understand how a virus works, that’s the first step towards stopping it,” said Prof. Gregory Voth, whose team created the model published in Biophysical Journal. “Each thing you know about the virus’s life cycle and composition is a vulnerability point where you can hit it.”
You can’t assume you can look at individual parts in isolation. Viruses are more than just the sum of their parts
Gregory Voth
Voth and his team drew on their previous experience to find the most important characteristics of each individual component of the virus, and drop the “less important” information to make a computational model that is comprehensive but still feasible to run on a computer. This technique is called coarse-graining, which Voth and his students have helped to pioneer.
The simplified framework helps address a key issue in health research: Even though a virus is one of the simpler biological entities, computational modeling is still a major challenge—especially if you want to model any of a virus’s interactions with its host’s body, which would mean representing billions of atoms. “You could try running an atom-level model of the actual entire virus, but computationally it would bog you down immediately,” Voth said. “You might be able to manage it long enough to model, say, a few hundred nanoseconds worth of movement, but that’s not really long enough to find out the most useful information.” Thus, many researchers have focused on creating models of individual proteins of the virus. But Voth said that while this segmented process has its uses, it also misses part of the larger picture.
“The virus itself is a holistic thing,” said Voth, a computational scientist and the Haig P. Papazian Distinguished Service Professor of Chemistry. “In my opinion, you can’t assume you can look at individual parts in isolation. Viruses are more than just the sum of their parts.”
Recommended article

Video • Computer model vs. Covid-19
Discovering new weaknesses of SARS-CoV-2 through bioinformatics
The search for antiviral agents against the new coronavirus is in full swing. Using a novel approach, Bioinformaticians have now discovered a weakness in the virus that could be exploited for drug development. In a computer model, the scientists discovered a human enzyme that is crucial for the virus. When it was switched off in the model, the virus could no longer multiply.
Voth said his lab has been working for years to model other viruses, such as HIV. One of the lessons they’ve learned is that multiple parts of the virus work in cooperation. For example, scientists might investigate a drug that binds to the spike proteins on the virus surface to prevent them from attaching to the host’s cells. “One of the main things you might want to know is, do you need to dose every spike protein for it to work? If not, how low a percentage can you get away with?” Voth said. “This is a key question when you’re trying to create drugs or antibodies, and it’s something you can best understand by looking at the entire virus.” The model also provides a framework into which scientists can integrate additional information about the SARS-CoV-2 virus as soon as new discoveries are made.
Voth hopes that the model will prove useful for coronavirus drug design as well as understanding mutations that may arise, such as the one recently detected in the U.K. Anyone can download the model and use it for their research. “Making a multiscale model of the whole virus and integrating all this information rapidly is a big technological step forward,” Voth said. “I’m really proud of my lab. We did it in record time, really—just a few months. If there is any upside to this pandemic, I hope that it advances our tools to fight viruses beyond Covid-19—like influenza, HIV and any new coronaviruses that arise in the future.”
Source: University of Chicago
12.01.2021