


Image source: Adobe Stock/alphaspirit
News • 'Synthetic origin' theory of SARS-CoV-2 debunked
Was the coronavirus engineered in a lab? Experts cast doubt on new analysis
The origin of SARS-CoV-2 remains unresolved. In a non-peer-reviewed preprint, three authors present analyses that, according to their interpretation, suggest a "synthetic emergence" of SARS-CoV-2 and its release in the context of a "laboratory accident". Experts from the University of Würzburg and the University Hospital have reviewed the preprint on the origin of SARS-CoV-2.
In summary, the analyses presented in the study do not provide sufficient evidence for the authors’ conclusion that SARS-CoV-2 is of synthetic origin.
In the non-peer-reviewed preprint published on BioRxiv, the authors present analyses that, according to their interpretation, suggest a "synthetic emergence" of SARS-CoV-2 and its release in the context of a "laboratory accident".
The core message of the preprint is that the genome of SARS-CoV-2 has an "abnormal pattern" of recognition sites for certain restriction enzymes (BsaI and BsmBI) and therefore is highly unlikely to have arisen by natural evolution. Based on statistical analyses, the authors conclude that this pattern most likely arose as a "fingerprint" in the SARS-CoV-2 genome during the establishment of a reverse genetics system for the (most likely bat-derived SARS-CoV-2 predecessor) in a research laboratory.
Restriction enzymes such as BsaI and BsmBI are used to clone genomes of coronaviruses isolated from animals such as bats and render them accessible for systematic research. Such so-called reverse genetics systems are of great importance for virological research. They allow the genetic conservation of otherwise mutation-prone viruses as well as the investigation of individual viral gene functions. Using these tools, coronavirus genomes, which are relatively large at just under 30,000 nucleotides, can be cloned and modified in bacteria in 5-8 small subfragments. For the cloning of the various fragments, individual bases of the viral genome occasionally have to be exchanged in order to insert the restriction sites necessary for cloning, or to remove unwanted restriction sites. Subsequently, the individual DNA fragments can be re-assembled to give rise to a complete viral genome.
The experts Florian Erhard, Oliver Kurzai and Lars Dölken from the Institute of Virology and Immune Biology and the Institute for Hygiene and Microbiology at the University of Würzburg, have subjected the preprint to scientific evaluation. Their main findings are:
1. Contrary to the authors' claim, the restriction site pattern of SARS-CoV-2 may well have arisen naturally - similar patterns are also found in coronaviruses closely related to SARS-CoV-2
Some known coronaviruses closely related to SARS-CoV-2 were obviously not included in the analyses performed in the preprint
Florian Erhard, Oliver Kurzai, Lars Dölken
All 5 restriction sites (BsmBI (n=3) and BsaI (n=2)) central to the analyses in the preprint are also commonly found in closely related coronaviruses. Thus, the existence of these 5 sites in the SARS-CoV-2 genome can be explained without human manipulation. Some known coronaviruses closely related to SARS-CoV-2 were obviously not included in the analyses performed in the preprint.
The authors of the preprint further argue that restriction sites in the SARS-CoV-2 genome that are unfavorable for genetic work are absent and presumably have been artificially altered ("deleted"). However, while many other coronaviruses actually show significantly more restriction sites for the two restriction enzymes analyzed, some closely related bat coronaviruses such as BANAL-20-103 and BANAL-116 also show only 5 and 7 restriction sites, respectively, with similarly sized genome fragments.
2. The position of the two BsaI restriction sites in the region of the S gene does not indicate genetic manipulation of the SARS-CoV-2 genome
The Spike protein of coronaviruses is of particular interest because it determines whether human cells can be infected or not. For reverse genetics models, it was therefore of particular interest for researchers to be able to exchange or modify the Spike protein coding region of coronavirus genomes. As shown by the authors, the two restriction sites for BsaI could be used to easily manipulate the most important part of the Spike protein of SARS-CoV-2, namely the receptor-binding domain (RBD) and the furin cleavage sites (FCS). For no other coronavirus isolate does this appear to be so easily possible with BsaI, since either the appropriate restriction sites are missing or additional sites would lead to additional unwanted fragments upon digestion with BsaI.
Recommended article
Article • Covid-19
Coronavirus update
Years after the first outbreak and spread of coronavirus SARS-CoV-2, its impact can still be felt in everyday life. Keep up-to-date with the latest research news, political developments, and background information on Covid-19.
The authors argue that this suggests that the SARS-CoV-2 genome has been optimized for easy replacement and manipulation of the most important parts of the Spike protein. In fact, the combination of BsaI and BsmBI has been used in the past by research groups in Wuhan to clone coronavirus genomes from bats and perform so-called gain-of-function experiments. However, in this case, the two BsaI restriction sites were each positioned to allow the exchange of the entire Spike protein. This is, however, not possible for SARS-CoV-2. If the two BsaI restriction sites had been in exactly the same positions as in previously published reverse genetics models, this would indeed have provided strong evidence for human manipulation. The two observed BsaI restriction sites are, however, also frequently found in closely related coronaviruses of SARS-CoV-2. It is noteworthy, however, that these coronaviruses usually possess at least one further BsaI site, which would have to be eliminated (i.e., mutated). Given the high mutation rate of circulating SARS-CoV-2 variants, one would also expect that artificially inserted synonymous (wobble) mutations, inserted to create or eliminate defined restriction sites, would disappear and artificially eliminated ones would reappear over the course of the more than two-year pandemic. In fact, however, the Omicron variants still have the same interface distribution pattern as the original Wuhan virus.
3. The statistical analyses of the paper on the distribution of restriction sites are flawed or incomplete in important respects
The analysis of a single, selectively chosen combination of two restriction enzymes (here: BsaI and BsmBI) is not suitable to prove human intervention
Florian Erhard, Oliver Kurzai, Lars Dölken
The combination of BsaI and BsmBI analyzed in the preprint is indeed not suitable for the vast majority of coronaviruses to dissect their genomes into an appropriate number of fragments (5 to 7) of suitable size (<8000 nucleotides) (Fig. 3C in the preprint; see below). However, as our own analyses showed, this is easily possible with other similar restriction enzymes. With the algorithms used and comprehensibly documented in the preprint, it can be shown that an appropriately suitable combination of restriction enzymes can be found for virtually any coronavirus genome. The analysis of a single, selectively chosen combination of two restriction enzymes (here: BsaI and BsmBI) is not suitable to prove human intervention. If one analyzes only one combination of two restriction enzymes suitable for a particular virus, it is predictable that this combination will be significantly less suitable for other virus isolates to construct a reverse genetics model. This leads to the authors' misinterpretation that the virus, in this case SARS-CoV-2, for which the selection of the analyzed combination of restriction endonucleases was optimized, is not of natural origin.
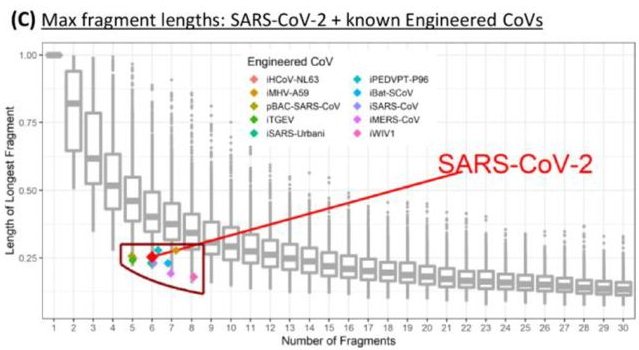
Image source: Bruttel et al., bioRxiv preprint 2022 (CC BY-NC-ND 4.0)
4. The analyses on the in silico evolution of two closely related coronaviruses with the aim of obtaining a restriction pattern comparable to that of SARS-CoV-2 (preprint Fig. 4) are not convincing
The assumption of purely random mutations in a viral genome is not valid, since most mutations disrupt or destroy the amino acid sequence of the viral proteins and are thus under selective pressure. In addition, the authors would also have had to analyze all acceptable combinations of restriction enzymes in this case as well.
The calculation described in the preprint of the probability for natural evolution of the observed restriction site pattern of SARS-CoV-2 is flawed. For this purpose, the authors combine the probabilities for a total of five different criteria. However, these are neither independent of each other, nor is the method used to combine these probability values appropriate. Furthermore, each individual probability calculated is affected by the same potential sources of error as listed above.
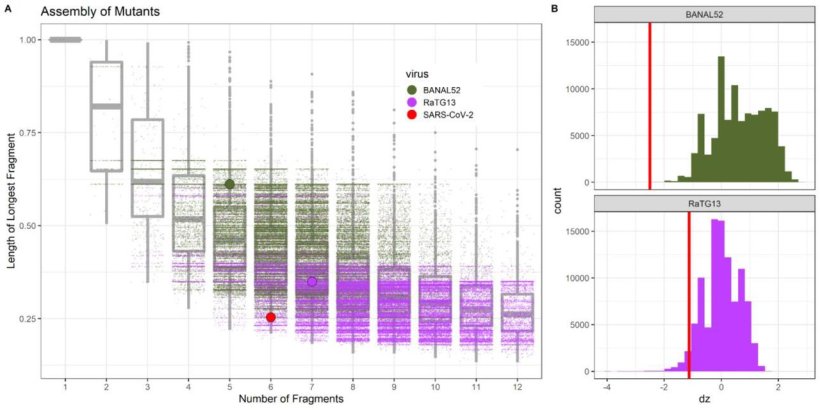
Image source: Bruttel et al., bioRxiv preprint 2022 (CC BY-NC-ND 4.0)
Conclusion
In their preprint, the authors present statistical analyses of the genome sequence of SARS-CoV-2 from which they conclude a synthetic origin of SARS-CoV-2. The preprint is carefully prepared and meets basic scientific requirements, particularly with respect to a sound and transparent presentation of the methodology used. However, the analyses presented in the preprint show considerable methodological weaknesses. As a result, the authors' main conclusions do not stand up to scientific scrutiny or result from over-interpretation of their analyses. In contrast to the statements formulated in the preprint, the pattern of restriction sites found in the genome of SARS-CoV-2 does not suggest genetic manipulation with the claimed probability. In summary, the analyses presented in the study do not provide sufficient evidence for the authors’ conclusion that SARS-CoV-2 is of synthetic origin. The origin of SARS-CoV-2 thus remains unresolved.
Source: University Hospital Würzburg
28.10.2022


