Source: University of Cambridge
News • COVID-19 infections
Genetic network analysis provides 'snapshot' of pandemic origins
Researchers from Cambridge, UK, and Germany have reconstructed the early “evolutionary paths” of COVID-19 in humans – as infection spread from Wuhan out to Europe and North America – using genetic network techniques.
By analysing the first 160 complete virus genomes to be sequenced from human patients, the scientists have mapped some of the original spread of the new coronavirus through its mutations, which creates different viral lineages. “There are too many rapid mutations to neatly trace a COVID-19 family tree. We used a mathematical network algorithm to visualise all the plausible trees simultaneously,” said geneticist Dr Peter Forster, lead author from the University of Cambridge. These techniques are mostly known for mapping the movements of prehistoric human populations through DNA. We think this is one of the first times they have been used to trace the infection routes of a coronavirus like COVID-19.”
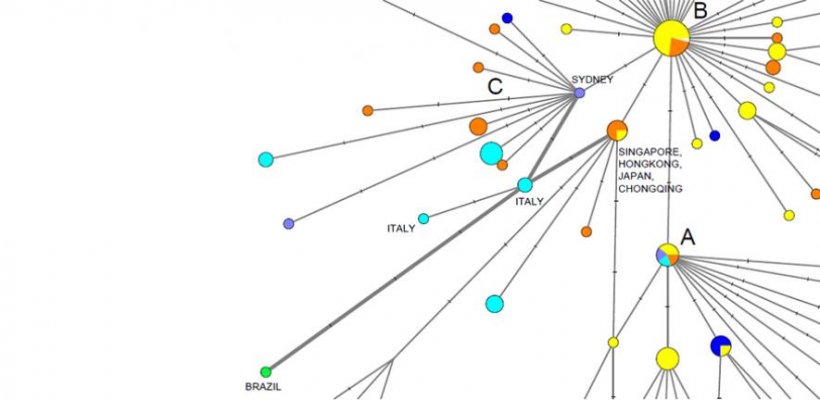
The team used data from virus genomes sampled from across the world between 24 December 2019 and 4 March 2020. The research revealed three distinct “variants” of COVID-19, consisting of clusters of closely related lineages, which they label ‘A’, ‘B’ and ‘C’.
Forster and colleagues found that the closest type of COVID-19 to the one discovered in bats – type ‘A’, the “original human virus genome” – was present in Wuhan, but surprisingly was not the city’s predominant virus type.
Versions of ‘A’ were seen in Chinese individuals, and Americans reported to have lived in Wuhan, and mutated versions of ‘A’ were found in patients from the US and Australia.
Wuhan’s major virus type, ‘B’, was prevalent in patients from across East Asia. However, the variant didn’t travel much beyond the region without further mutations – implying a "founder event" in Wuhan, or “resistance” against this type of COVID-19 outside East Asia, say researchers.
The ‘C’ variant is the major European type, found in early patients from France, Italy, Sweden and England. It is absent from the study’s Chinese mainland sample, but seen in Singapore, Hong Kong and South Korea.
The new analysis also suggests that one of the earliest introductions of the virus into Italy came via the first documented German infection on January 27, and that another early Italian infection route was related to a “Singapore cluster”.
Importantly, the researchers say that their genetic networking techniques accurately traced established infection routes: the mutations and viral lineages joined the dots between known cases.
Credit: NIAID-RML
As such, the scientists argue that these “phylogenetic” methods could be applied to the very latest coronavirus genome sequencing to help predict future global hot spots of disease transmission and surge. “Phylogenetic network analysis has the potential to help identify undocumented COVID-19 infection sources, which can then be quarantined to contain further spread of the disease worldwide,” said Forster, a fellow of the McDonald Institute of Archaeological Research at Cambridge, as well as the University’s Institute of Continuing Education.
The findings are published today in the journal Proceedings of the National Academy of Sciences (PNAS). The software used in the study, as well as classifications for over 1,000 coronavirus genomes and counting, is available free at www.fluxus-technology.com.
Variant ‘A’, most closely related to the virus found in both bats and pangolins, is described as “the root of the outbreak” by researchers. Type ‘B’ is derived from ‘A’, separated by two mutations, then ‘C’ is in turn a “daughter” of ‘B’.
Researchers say the localisation of the ‘B’ variant to East Asia could result from a “founder effect”: a genetic bottleneck that occurs when, in the case of a virus, a new type is established from a small, isolated group of infections.
Forster argues that there is another explanation worth considering. “The Wuhan B-type virus could be immunologically or environmentally adapted to a large section of the East Asian population. It may need to mutate to overcome resistance outside East Asia. We seem to see a slower mutation rate in East Asia than elsewhere, in this initial phase.”
He added: “The viral network we have detailed is a snapshot of the early stages of an epidemic, before the evolutionary paths of COVID-19 become obscured by vast numbers of mutations. It’s like catching an incipient supernova in the act.”
Since the PNAS study was conducted, the research team has extended its analysis to 1,001 viral genomes. While yet to be peer-reviewed, Forster says the latest work suggests that the first infection and spread among humans of COVID-19 occurred between mid-September and early December.
The phylogenetic network methods used by researchers – allowing the visualisation of hundreds of evolutionary trees simultaneously in one simple graph – were pioneered in New Zealand in 1979, then developed by German mathematicians in the 1990s.
Source: University of Cambridge
14.04.2020