Image source: Adobe Stock/lucadp
News • Genetics & Neuroscience
Research investigates the convergence of schizophrenia risk genes in brain coexpression networks
Johns Hopkins researchers, along with colleagues in Italy, have published a study that looks into the genetic mechanisms behind the development of schizophrenia.
The study that has been published in Science Advances focuses on the convergence of schizophrenia risk genes in brain coexpression networks in the postmortem human prefrontal cortex, hippocampus, caudate nucleus, and dentate gyrus granule cells, grouped by specific age brackets in 833 samples, 186 of which were sourced from schizophrenia-diagnosed patients.
The results support an early involvement in the prefrontal brain with gene expression signatures underlying schizophrenia and reveal a dynamic interplay of regions in which age bracketing explains more variance in schizophrenia risk compared to analyzing all age brackets together.
Genetic architecture of schizophrenia
The genetic architecture of schizophrenia was found to include shifting coexpressed gene set patterns across brain regions and time, potentially explaining shifting observed clinical presentations in patients. Specifically, there was a large convergence of risk genes expressed in the dorsolateral prefrontal cortex of juveniles.
The dorsolateral prefrontal cortex (DLPFC) is an area of the brain where many essential activities occur, such as active, conscious decision-making, working memory, outcome predictions, impulse inhibition and reasoned thoughts—collectively known as executive functions. It is also an important node in the cognitive selection of sensory information.
Using past studies to identify 28 genes consistently found partnered in modules enriched for schizophrenia risk genes in DLPFC, researchers found 23 previously recorded in schizophrenia studies but with unidentified associations with risk genes. These co-expressed genes may have eluded past researchers because of the age-dependent way they are expressed.
An example given in the paper illustrates how schizophrenia risk modules enriched for a transcription factor, prioritized in genetic associations with schizophrenia, are found in the perinatal and juvenile DLPFC but not later and so go missing from data that is not age specific.
The study design
In this study, four hypotheses have been evaluated:
(i) Age-parsed networks in neurotypical brains explain more SCZ genetic risk than the same data not age-parsed.
(ii) The course of SCZ enrichment differs between NCs and patients with SCZ.
(iii) There is a molecular environment to SCZ risk that can be identified by consensus between networks in terms of genes coexpressed with SCZ risk genes.
(iv) The consensus genetic environment surrounding SCZ risk genes can be reproduced in vitro to perform cell system studies of coexpression networks relevant to SCZ. CN, caudate nucleus; HP, hippocampus; MAGMA, multimarker analysis of genomic annotation; SCZ, schizophrenia.
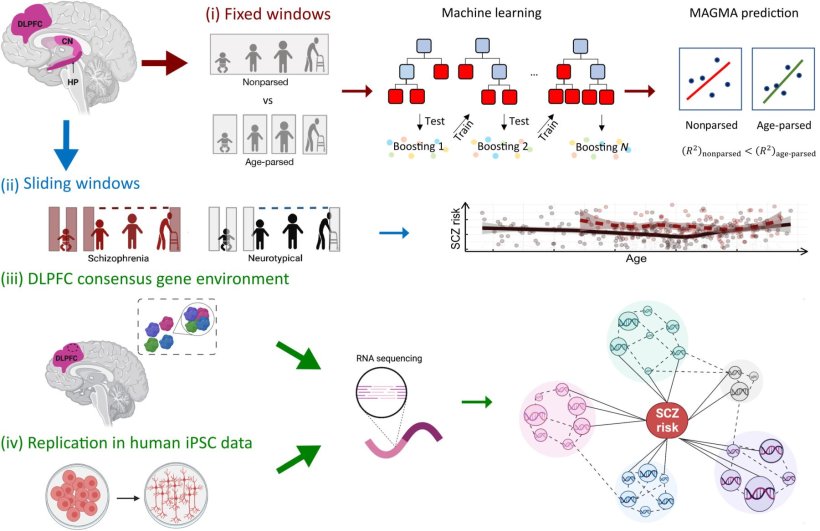
Image source: Science Advances (2023). DOI: 10.1126/sciadv.ade2812
Identification of mutation-intolerant consensus genes
Researchers report that the most unexpected and novel result of the study is the identification of mutation-intolerant consensus genes that are especially likely to be coexpressed with schizophrenia risk genes. This set of genes was coexpressed with risk genes in most networks examined, regardless of differences across datasets, preprocessing pipelines, and parameter settings.
When merging data of modules containing these consensus genes, the results showed strong enrichment for known schizophrenia risk genes, which was not found when merging modules without consensus genes. Gene ontologies for the consensus gene set and their modules emphasized neurons, synapses, and ion transport. From a chromosomal view, these consensus genes are not clustered, ruling out locus-related artifacts.
The novel way in which the study set out to explore the evolution of schizophrenia by age groups allowed the researchers to observe correlations that were present and yet missed in past studies. Having a better perspective of commonly coexpressed genes provides a more complete picture of the molecular environment in which the disease is taking place and moves us closer to identifying the mechanisms at work. It could also help clinicians better understand the disparate manifestations observed in patients over time.
Source: Science X Network
20.04.2023