Credit: Salk Institute
News • From foe to friend?
Some Alzheimer’s plaques may be protective, not destructive
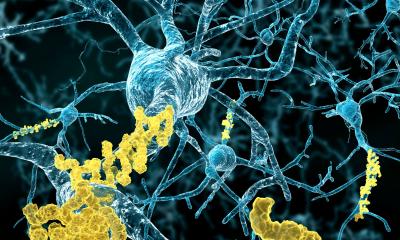
One of the characteristic hallmarks of Alzheimer’s disease (AD) is the buildup of amyloid-beta plaques in the brain. Most therapies designed to treat AD target these plaques, but they’ve largely failed in clinical trials. New research by scientists at the Salk Institute upends conventional views of the origin of one prevalent type of plaque, indicating a reason why treatments have been unsuccessful.
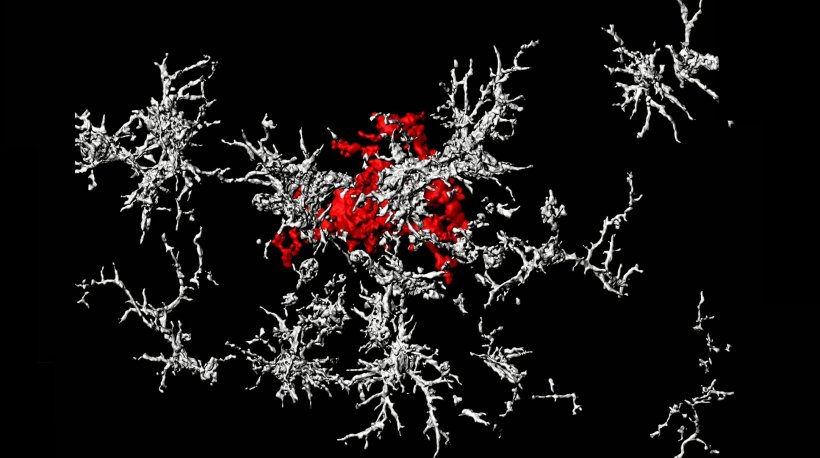
The traditional view holds that the brain’s trash-clearing immune cells, called microglia, inhibit the growth of plaques by “eating” them. The Salk scientists show instead that microglia promote the formation of dense-core plaques, and that this action sweeps wispy plaque material away from neurons, where it causes cell death. The research, which was published in Nature Immunology, suggests that dense-core plaques play a protective role, so treatments to destroy them may do more harm than good.

Credit: Salk Institute
“We show that dense-core plaques don’t form spontaneously. We believe they’re built by microglia as a defense mechanism, so they may be best left alone,” says Greg Lemke, a professor in Salk’s Molecular Neurobiology Laboratory. “There are various efforts to get the FDA to approve antibodies whose main clinical effect is reducing dense-core plaque formation, but we make the argument that breaking up the plaque may be doing more damage.”
Alzheimer’s disease is a neurological condition that results in memory loss, impairment of thinking, and behavioral changes, which worsen as we age. The disease seems to be caused by abnormal proteins aggregating between brain cells to form the hallmark plaques, which interrupt activity that keeps the cells alive. There are numerous forms of plaque, but the two most prevalent are characterized as “diffuse” and “dense-core.” Diffuse plaques are loosely organized, amorphous clouds. Dense-core plaques have a compact center surrounded by a halo. Scientists have generally believed that both types of plaque form spontaneously from excess production of a precursor molecule called amyloid precursor protein (APP). But, according to the new study, it is actually microglia that form dense-core plaques from diffuse amyloid-beta fibrils, as part of their cellular cleanup.
This builds on a 2016 discovery by the Lemke lab, which determined that when a brain cell dies, a fatty molecule flips from the inside to the outside of the cell, signaling, “I’m dead, eat me.” Microglia, via surface proteins called TAM receptors, then engulf, or “eat” the dead cell, with the help of an intermediary molecule called Gas6. Without TAM receptors and Gas6, microglia cannot connect to dead cells and consume them.
The team’s current work shows that it’s not only dead cells that exhibit the eat-me signal and Gas6: So do the amyloid plaques prevalent in Alzheimer’s disease. Using animal models, the researchers were able to demonstrate experimentally for the first time that microglia with TAM receptors eat amyloid plaques via the eat-me signal and Gas6. In mice engineered to lack TAM receptors, the microglia were unable to perform this function. Digging deeper, they traced the dense-core plaques using live imaging. Much to their surprise, the team discovered that after a microglial cell eats a diffuse plaque, it transfers the engulfed amyloid-beta to a highly acidic compartment and converts it into a highly compacted aggregate that is then transferred to a dense-core plaque. The researchers propose that this is a beneficial mechanism, organizing diffuse into dense-core plaque and clearing the intercellular environment of debris.

Credit: Salk Institute
“Our research seems to show that when there are fewer dense-core plaques, there seem to be more detrimental effects,” says Youtong Huang, first author on the paper. “With more-diffuse plaques, there’s an abundance of dystrophic neurites, a proxy for neuronal damage. I don’t think there’s a distinct clinical decision on which form of plaque is more or less detrimental, but through our research, we seem to find that dense-core plaques are a bit more benign.”
Their findings suggest new ways of developing a treatment for Alzheimer’s disease, such as boosting expression of TAM receptors on microglia to accelerate dense-core plaque formation. The team would like to conduct cognitive studies to see if increasing the activity of microglial TAM receptors would alleviate the effects of AD.
Lemke, who holds the Françoise Gilot-Salk Chair, believes that the current failure rate of most Alzheimer’s drug trials is about to end. “Some people are saying that the relative failure of trials that bust up dense-core plaques refutes the idea that amyloid-beta is a bad thing in the brain,” says Lemke. “But we argue that amyloid-beta is still clearly a bad thing; it’s just that you’ve got to ask whether dense-core plaques are a bad thing.” Lemke suggests that scientists looking for a cure for Alzheimer’s should stop trying to focus on breaking up dense-core plaques and start looking at treatments that either reduce the production of amyloid-beta in the first place or therapies that facilitate transport of amyloid-beta out of the brain altogether.
Source: Salk Institute
16.04.2021