Gene Mutation
Researchers identify cause of hereditary skeletal muscle disorder
An international research team from the University Hospital of Munich (LMU), Newcastle University and the University of Liverpool has identified a gene mutation that causes a hereditary skeletal muscle disorder. The study is published in the "American Journal of Human Genetics".
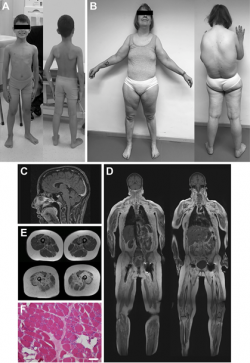
The starting point for this discovery was a family in which two of six children dad been living with delayed motor development and muscle weakness since birth. In addition to skeletal muscles, eyes and brain seemed to be affected as well: Both patients developed early-onset cataracts and had mild mental retardation. The scientists analysed the whole exome of the patients, that is to say all regions of the DNA that encode proteins. This analysis resulted in a suspicious variant in the gene INPP5K, which appeared to be related to the manifestation of the disease. Definite proof came from genetic studies involving further unrelated patients with the same disorder: Together with colleagues at home and abroad, the research team came across INPP5K mutations in seven additional families.
In order to understand the mechanism of the disorder, the researchers investigated the function of INPP5K in zebrafish larvae. The loss of INPP5K caused defective development of skeletal muscles and eyes, replicating essential features of the human disease. The INPP5K gene provides the blueprint for making an enzyme that controls the turnover of phosphoinositides, small lipids that are involved in the regulation of a variety of cell and organ functions. Further experiments indeed showed that most disease-related INPP5K mutations resulted in strongly impaired enzyme function. The association between abnormal phosphoinositide metabolism and hereditary human diseases had already been demonstrated in earlier studies. However, mutations did not affect the INPP5K gene but were found in genes for other phosphoinositide-metabolizing enzymes.
The condition caused by INPP5K mutations is one of the rare diseases. "It was only thanks to a worldwide cooperation with doctors and geneticists, that we were able to identify several affected families and confirm our initial suspicion," says Professor Jan Senderek from the Friedrich Baur Institute of the University Hospital of Munich (LMU) and last author of the publication. Although the results of the study do not yet establish a causative therapy, the identification of the gene defect is expected to pave the way for the search for new treatment options. And already now, the results are of practical importance, as Professor Senderek notes: "The discovery of the cause of the disease enables targeted genetic diagnostics and counselling of affected families and thus contributes to improved care of patients with this rare disease."
Source: Friedrich-Baur-Institut an der Neurologischen Klinik und Poliklinik, Klinikum der Universität München (LMU)
27.02.2017