

Heart failure
Researchers identify 'missing culprit'
Working with lab animals and human heart cells, scientists from Johns Hopkins and other institutions have identified what they describe as "the long-sought culprit" in the mystery behind a cell-signaling breakdown that triggers heart failure.
The condition, which affects nearly 6 million Americans and 23 million people worldwide, is marked by progressive weakening and stiffening of the heart muscle and the organ's gradual loss of blood-pumping ability.
The research results, described March 18 in the journal Nature, reveal that an enzyme called PDE-9 interferes with the body's natural "braking" system needed to neutralize stress on the heart. The experiments demonstrate that the enzyme wreaks mischief by gobbling up a signaling molecule, cGMP, which normally stimulates the production of a heart-protective protein called PKG, known to shield the heart muscle from the ravages of disease-causing stress, such as long-standing high blood pressure.
Naturally found in the gut, kidneys and brain, PDE-9 is already a prime suspect in neurodegenerative conditions such as Alzheimer's, the researchers say. But the new study shows the enzyme's footprints are also present in heart cells and markedly elevated in patients with heart failure -- evidence that PDE-9 is a multitasking "offender" and a key instigator of heart muscle demise, the researchers say.
To understand the enzyme's role, the scientists exploited the knowledge that heart muscle health is safeguarded by two separate mechanisms, or signaling pathways. Activated by two different chemicals -- nitric oxide and natriuretic peptide -- each pathway produces cGMP, which in turn stimulates the all-important heart muscle protector PKG. Most cases of heart failure, the researchers say, are fueled by breakdowns in both.
"The existence of two separate pathways with overlapping but distinct functions is nature's insurance policy, a fail-safe redundancy to ensure that should one pathway falter, the other one can compensate and maintain heart muscle function," says senior investigator David Kass, M.D., professor of medicine at the Johns Hopkins University School of Medicine and its Heart and Vascular Institute.
Nearly a decade ago, a team led by Kass identified the culprit responsible for breakdown in one of the signaling pathways, an enzyme called PDE-5 -- also known to cause erectile dysfunction -- and ever since then scientists have searched for the second "offender" that causes glitches in the other pathway. The discovery of PDE-9 provides that long-sought "break in the case," the team says.
"Like a play with multiple characters, heart muscle function is the result of a complex but perfectly synchronized interaction of several proteins, enzymes and hormones," says lead investigator Dong Lee, M.S., Ph.D., a cardiology research associate at the Johns Hopkins University School of Medicine. "Our findings reveal that, like two subplots that converge in the end of the play, PDE-5 and PDE-9 are independent rogue operators, each leading to heart muscle damage but doing so through different means."
Kass' earlier studies showed that PDE-5, like its newly identified accomplice PDE-9, inflicts damage by feeding on heart-protective cGMP and PKG. But the new findings reveal an important difference -- PDE-9 has an appetite for the form of cGMP stimulated by the second signaling pathway.
In other words, Kass says, too much PDE-9 can interfere with the second of the two heart-protective systems by speeding up the breakdown of cGMP, which in turn reduces levels of PKG, rendering heart cells prone to malfunction and the heart muscle vulnerable to scarring and damage.
The research team also notes that heart failure treatments blocking the activity of PDE-9 may be right around the corner, with drugs that inhibit PDE-9 already being tested for use in people with Alzheimer's disease.
In the current study, such PDE-9 blockers not only stopped heart muscle enlargement and scarring in mice with heart failure, but they nearly reversed the effects of the disease.
"We believe the identification of PDE-9 puts us on the cusp of creating precision therapies that target the second pathway or developing combined therapies that avert glitches in both pathways," Kass says.
The new findings could be even more relevant for the nearly one-half of all heart failure patients with an especially recalcitrant form of the disease known as heart failure with preserved ejection fraction -- a treatment-defying condition in which the heart appears to pump normally but is, in fact, scarred and hardened. Heart cells obtained from people with this form of the disease showed PDE-9 levels six times higher than normal hearts, suggesting that people with that form of the disease have far less heart-protective PKG, the researchers note.
In one set of experiments, the researchers chemically stimulated human heart cells and cells from newborn mice to mimic the effects of heart failure. The cells ballooned beyond their normal size, but when treated a PDE-9 antidote, the cells returned to near normal.
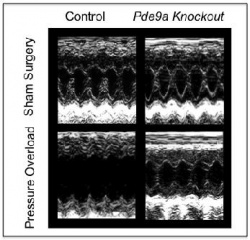
In another test, the scientists created mice genetically incapable of making PDE-9 and compared their heart cells and overall organ function to mice with intact PDE-9 genes. The heart cells of mice lacking PDE-9 had higher cGMP levels than the cells of mice with intact PDE-9. Next, mice in both groups underwent surgical narrowing of the aorta, the body's main blood vessel, a procedure intended to create extra workload for the heart and induce heart failure. Mice lacking the PDE-9 gene fared much better. Their hearts had far less scarring, muscle thickening and dilation than the hearts of mice with intact PDE-9.
In a third experiment, the scientists divided mice with surgically induced heart failure into three groups. One group received treatment with a PDE-9 blocking drug, another was treated with the PDE-5 blocker sildenafil and a third was given placebo.
After four weeks of therapy, mice treated with placebo developed full-blown heart failure, while mice treated with either PDE-9 or PDE-5 blockers showed marked improvements in heart muscle function and size, their hearts' pumping ability reverting to near normal.
To compare the effects of PDE-5 and PDE-9 inhibitors on heart function, the researchers then fed half the mice with a chemical that turned off the heart-protective signaling pathway regulated by PDE-5. In that group, treatment with a PDE-5 inhibitor made no difference. However, when given a PDE-9 blocker, the animals' hearts showed notable improvement. This finding, the research team says, illustrates the notion that heart muscle failure is fueled by flaws in two separate signaling cascades, each one regulated by a different enzyme and benefiting from different treatment.
"In practical terms, this affirms that preserving the function in one pathway can avert clinical disease, even if the other one goes bad," Kass says.
Source: Press release Johns Hopkins Medicine
20.03.2015

