Computational modeling
Analyzing genetic tree sheds light on disease outbreaks
Scientists have a new tool for unraveling the mysteries of how diseases such as HIV move through a population, thanks to insights into phylogenetics, the creation of an organism’s genetic tree and evolutionary relationships.
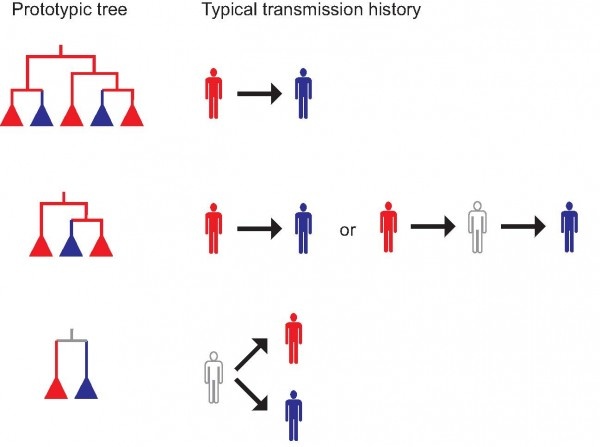
“It turns out that three different types of transmission histories are possible between two persons who might have infected each other,” said Thomas Leitner of Los Alamos National Laboratory, the corresponding author of a new paper out this week in the Proceedings of the National Academy of Sciences. “Using phylogenetic inference in the epidemiological investigations of HIV transmission, we’ve determined that between two sampled, potentially epidemiologically linked persons, we can now evaluate the possibility that an unsampled intermediary or common source existed, even without a sample from that individual.”
Like a detective inferring the existence of an unseen actor in a sequence of events, the Los Alamos team used computational phylogenetic analysis to examine how strains of HIV, both in computer modeling and compared with real-life case studies, would be transmitted.
The team’s research has broad implications. “The inference of donor-recipient relationships we describe here is not restricted to HIV transmissions; it applies to all situations when an original population seeds a new population with a restricted random draw (a bottleneck) of individuals. We use HIV transmission to illustrate the effects because it helps trace contacts among people and untangle investigations into outbreaks. Also, statistical guidelines are needed for interpreting phylogenetic results in court.”
Phylogenetic inference of who infected whom has great value in epidemiological investigations, the authors point out, because it should explain how transmission(s) occurred. Until now, however, there has not been a systematic evaluation of which phylogeny to expect from different transmission histories, and thus interpreting the meaning of an observed phylogeny has remained elusive.
“Previously it was thought that it would be impossible to say who infected whom and whether there were unsampled intermediary links in an alleged transmission, or if both persons were infected by an unsampled/unknown third party. We show that this is now possible in many cases,” Leitner said. “This will have large impact on future epidemiological investigations, including forensics and outbreak investigations."
In the paper, the team showed that certain types of phylogenies associate with different transmission histories, which may make it possible to exclude possible intermediary links or identify cases where a common source was likely but not sampled. “Our systematic classification and evaluation of expected topologies should make future interpretation of phylogenetic results in epidemiological investigations more objective and informative,” Leitner said.
Source: Los Alamos National Laboratory
09.03.2016